一种碳纳米片材料及其制备和在钠离子电池中的应用的制作方法
本发明涉及一种碳纳米片材料的制备方法及其在钠离子电池中的应用,属于钠离子电池领域。
背景技术:
伴随煤炭、石油、天然气等传统能源的逐渐减少以及日益严峻的环境问题,小型分离移动电源需求呈现出爆炸式增长趋势,以锂电池为代表的各种可充电的化学电源越来越受到重视。然而,由于锂在地壳中的元素含量相对较少,锂的提取和回收困难,因此有必要开发新型的电池体系。
钠离子电池是近年来快速发展的高性能储能体系。钠在自然界中的储量非常丰富,约占地壳的2.74%,并且广泛分布,有效地降低了成本。同时钠与锂同为第i主族元素,两者具有相似的化学特性。因此,钠离子电池具有和锂离子电池类似的脱嵌机制被认为是大规模储能领域的理想选择。
目前,在二次电池领域,基于材料开发成本以及应用前景的考虑,研究较多的钠离子负极材料主要是各种碳基材料,如石墨、石墨烯、无定型碳等,碳基材料的电化学性能与各自结构有关。传统碳基材料导电性差、性能循环稳定性差,容量衰减快,极大限制了其在钠离子电池中应用。因此,如何提高碳材料作为钠离子电池负极的比容量和长期循环稳定性能,成为限制碳材料作为钠离子电池负极材料大规模应用的关键问题。
技术实现要素:
针对现有钠离子电池电极材料存在的缺陷,本发明的提供一种碳纳米片材料的制备方法,该方法工艺简单、重复性好、成本低廉、环境友好,可控度高,易于实现工业化。
本发明的第二目的在于提供所述的制备方法制得的碳纳米片材料。
本发明的第三目的在于提供制得的碳纳米片材料的应用,旨在提升制得的钠离子电池的电学性能,例如提高其比容量和长期循环稳定性能。
一种碳纳米片材料的制备方法,包括以下步骤:
步骤(1):将包含过渡金属盐、聚乙二醇、碱的水溶液进行水热反应,制得金属氧化物纳米片;
步骤(2):将所述的金属氧化物纳米片、多巴胺和氨基酸置于tris缓冲液中、混合制得前驱体;将所述前驱体在900~1200℃下热处理(本发明也称为碳化),随后经洗涤、干燥,即得。
本发明中,在所述的水热条件下有利于得到更适宜制备碳纳米片的金属氧化物纳米片;随后再配合多巴胺的自聚合性和在金属表面易成膜的性质,在金属氧化物纳米片的表面均匀聚合了有机碳源和氮源,无需水热反应过程,即可在在金属纳米片模板表面原位包覆有机层,最后再在所述的温度下碳化,可得到含氮量、层间距可控、性能优异的不定形多孔碳纳米片材料。
本发明的制备方法,可通过控制氨基酸以及多巴胺的用量,实现了对制得的碳纳米片材料的掺氮量的精准调控。通过本发明方法,可方便调控得到的材料的掺氮量,且在所述的水热反应以及热处理条件的协同,可制得层间距大,具有优良导电性,丰富的官能团,同时具有疏松多孔结构的碳纳米片材料,该材料用作钠离子电池负极材料,可表现出高的比容量、良好倍率性能和长循环稳定性能。
本发明制备方法;碳纳米片中氮含量可控,制备方便,适用于大规模工业化生产。本发明方法制备的碳纳米片氮掺杂含量丰富,孔道交联度高,反应活性位点丰富,层间距适中;作为钠离子电池负极材料,具有良好的电池传输动力学性能,氮原子的掺入显著地提高了材料导电性,大的层间距为钠离子嵌脱过程造成的体积膨胀提供了缓冲空间。
作为优选,所述的过渡金属盐为铁的水溶性盐、钴的水溶性盐、镍的水溶性盐、铜的水溶性盐中的至少一种。例如,所述的过渡金属盐为铁、钴、镍、铜的硝酸盐、氯化盐、醋酸盐等水溶性盐。
进一步优选,所述过渡金属盐为氯化铁,硝酸钴,乙酸镍,氯化铜中的至少一种,更优选为氯化铁。
所述的碱为碱金属的氢氧化物,优选为氢氧化钠。
作为优选,所述过渡金属盐与碱(以oh-计)的摩尔比为1~1000∶1。也即是,优选将水热反应的起始水溶液的ph控制在6.5~9。
进一步优选,所述过渡金属盐与碱(以oh-计)的摩尔比为10~50∶1。
所述的聚乙二醇优选的分子量为600~4000。
作为优选,所述过渡金属盐与聚乙二醇的摩尔比例为1~5∶1。
将包含所述比例的过渡金属盐与聚乙二醇的水溶液进行水热反应,制得金属氧化物纳米片;研究表明,合适的水热反应温度,有利于制得更适合碳纳米片制备的金属氧化物纳米片。
作为优选,水热反应温度为140~220℃。在优选的温度下,水热制得的金属氧化物纳米片形貌规整,孔道丰富,利于后续制备过程中多巴胺的附着。
作为优选,水热反应温度为150~190℃;更优选为160~180℃。在该优选的温度下,制得的金属纳米片更适合碳纳米片的制备。
在所述的水热条件,优选的水热反应时间为16~48h;进一步优选为16~24h。
优选的方案,所述二维金属氧化物纳米片置于溶有盐酸多巴胺的tris缓冲液中,随后加入氨基酸,多巴胺自聚在二维金属氧化物表面并吸附氨基酸,在金属纳米片表面形成规则的片状结构;即得到前驱体。
较优选的方案,所述氨基酸为甘氨酸、天冬氨酸、天冬酰胺、谷氨酸、赖氨酸、谷氨酰胺、丝氨酸、苏氨酸、半胱氨酸、组氨酸和精氨酸其中的一种或几种。
本发明可通过调节多巴胺与金属氧化物纳米片的比例,调控碳纳米片的孔容和比表面积,进而制得性能优异的碳纳米片材料。
较优选的方案,多巴胺与金属氧化物纳米片的摩尔比为1~100∶1。在该优选的范围下,得到的碳纳米片的材料的性能更优异。
进一步优选。多巴胺与金属氧化物纳米片的摩尔比为10~50:1。
本发明通过调节多巴胺与氨基酸的比例,调控氮的掺入量;还有助于获得较大的层间距;进而,进一步协同提升制得的碳纳米片的电学性能。
较优选的方案,多巴胺和氨基酸的摩尔比为1~5∶1。在该优选的比例下,得到的材料的电学性能更优异。
进一步优选,多巴胺和氨基酸的摩尔比为1~3∶1。
步骤(2)中,多巴胺在溶液中的浓度优选为1~100g/l。
tris缓冲液的ph优选为8~9。
本发明中,前驱体在保护气氛下热处理,所述的保护气氛例如为氮气、惰性气体气氛。
本发明研究发现,采用所述的前驱体,控制在所优选的温度下进行碳化,可制得层间距大,具有优良导电性,丰富的官能团,同时具有疏松多孔结构的碳纳米片材料。低于此温度下限,前驱体碳化不充分,得到的碳纳米片材料的导电性差;高于该温度上限时,得到的碳纳米片材料失去无定型结构向石墨材料转化,容量发挥差,衰减快。
进一步优选,碳化温度为1000~1100℃。在该优选的温度下保温碳化,得到的材料的导电性更好,材料的比容量高,循环性能更优异。
作为优选,以1~10℃/min升温至所述的热处理温度;并在所述的热处理温度下保温,优选的保温时间(本发明也称为热处理时间)为0.5~20h。
进一步优选,升温速率为1~3℃/min。
在所述的热处理温度下,优选的保温时间为2~10h。
本发明中,热处理得到碳化产物,将碳化产物进行洗涤,例如通过酸洗去除金属,制得所述的碳纳米片材料;洗涤得到的过渡金属盐回收、套用。采用酸洗后的产物再采用水洗至中性,洗涤可以反复进行,直至将金属全部去除。洗涤后的产物,置于50~80℃温度条件下,真空干燥8~12h。采用的酸溶液为本领域公知的稀酸溶液,采用稀酸和水反复交替洗涤,能将残留的金属纳米片及碳化过程中产生的杂质去除。采用的酸溶液如稀盐酸、稀硫酸、稀硝酸中的至少一种;所述酸溶液浓度一般在0.5mol/l左右。
本发明一种优选的碳纳米片材料的制备方法,在含过渡金属盐和聚乙二醇的溶液中滴加氢氧化钠溶液,再转移至反应釜中用水热法制备金属氧化物纳米片;将二维金属氧化物纳米片,多巴胺和氨基酸在tris缓冲液中混合制得前驱体;所述前驱体在900~1200℃下热处理后,洗涤,既得。
本发明的制备碳纳米片材料的方法包括以下具体步骤:
(1)将过渡金属盐在搅拌条件下充分溶解于去离子水和聚乙二醇的溶液中;缓慢滴加氢氧化钠溶液,搅拌均匀后,置于密封釜内保温16~48h,保温温度160~180℃,得到金属氧化物纳米片;溶液中过渡金属盐浓度为1~3mol/l,聚乙二醇浓度为0.01~100g/l;采用的氢氧化钠溶液浓度为1~3mol/l,过渡金属盐与氢氧化钠摩尔比为1~1000∶1;
(2)将上述将二维金属氧化物纳米片作为模板置于溶有盐酸多巴胺的tris缓冲液中,随后加入氨基酸静置4~24h,即得到前驱体。混合物中多巴胺浓度为1~100g/l,混合物中氨基酸浓度为0.2~100g/l,多巴胺与过渡金属纳米片的摩尔比为0.001~1∶1,多巴胺和氨基酸摩尔比为1~3∶1;
(3)将前驱体进行碳化,碳化温度为1000~1100℃,碳化时间为0.5~10h,升温速率为1~10℃/min;高温碳化产物采用稀酸溶液与水反复洗涤后,置于50~80℃温度条件下,真空干燥8~12h;得到含氮量12~33%,纳米片材料厚度在30~300nm,层间距在0.37~0.471nm的碳纳米片材料。
本发明还提供了一种所述的制备方法制得的碳纳米片材料,为掺氮碳的二维片状材料。
优选的方案,所述碳纳米片材料氮原子含量(原子个数百分含量)12~33%。在该优选的掺氮量下,该材料的储钠活性位点更丰富,导电性更好。
作为优选,所述碳纳米片材料氮原子含量12~29%;进一步优选为20~29%。
优选的方案,所述碳纳米片材料厚度在30~300nm,更优选为30-45nm;所述碳纳米片层间距在0.37~0.471nm;进一步优选为0.39~0.47。
所述碳纳米片材料具有丰富空隙,比表面积为200~800m2/g;进一步优选为450~800m2/g。
本发明方法制得的碳纳米片材料层间距大,储钠空间足够,具有良好的钠离子嵌入脱出能力。该材料还具有优良导电性,丰富的官能团,同时具有疏松多孔结构,作为钠离子电池负极,表现出高的比容量、良好倍率性能和长循环稳定性能。
本发明还提供了一种碳纳米片材料的应用,作为钠离子电池的负极材料,用于制备钠离子电池的负极。
钠离子电池负极的制备方法可采用现有方法,例如,将本发明得到的碳纳米片材料与导电剂和粘结剂混合,通过涂布法涂覆在铜箔集流体上,制得钠离子电池负极。
本发明制备的碳纳米片材料作为负极材料制备钠离子电池的方法及性能检测方法:称取上述碳纳米片材料,加入10wt.%superp作为导电剂,10wt.%羧甲基纤维素钠(cmc)作为粘结剂,经研磨充分之后加入少量去离子水混合形成均匀的黑色糊状浆料,将这些浆料涂覆在铜箔集流体上作为测试电极,以金属钠片作为对比电极组装成为扣式电池,其采用电解液体系为1mnaclo4/ec:dec(1:1)。测试循环性能所用充放电电流密度为100ma/g。
有益效果:
1)本发明的技术方案采用多巴胺做为有机碳源,所述的氨基酸为氮源,以二维金属氧化物纳米片为模板,经过高温碳化,最终制备了氮掺杂的碳纳米片材料,该方法采用原料廉价易得,制备工艺简单,重现性好,绿色环保,适合工业化生产。
2)本发明的技术方案制备的碳纳米片材料具有高的氮掺杂含量和合适的层间距,碳层之间丰富的官能团和氮原子掺入造成的缺陷为钠离子的储存提供了丰富的活性位点。多孔碳材料中大的碳层间距,可以有效促进钠离子在碳层间的嵌入和脱出过程,提高碳材料的比容量。此外,本材料比表面积大,机械强度高,导电性好,石墨化程度低。
3)本发明的技术方案制备的碳纳米片材料克服了现有掺杂原子碳材料杂原子分布不均的问题,均匀分布的官能团和氮原子引入的缺陷为钠离子的储存提供了丰富的活性位点,提供了供钠离子嵌脱过程中材料体积收缩膨胀的空间。
4)本发明的碳纳米片材料可用于制备具有高库伦效率、优异的倍率性能和长循环稳定性能的钠离子电池。
附图说明
【图1】为实施例1制得的碳纳米片材料料的sem扫面电镜图。
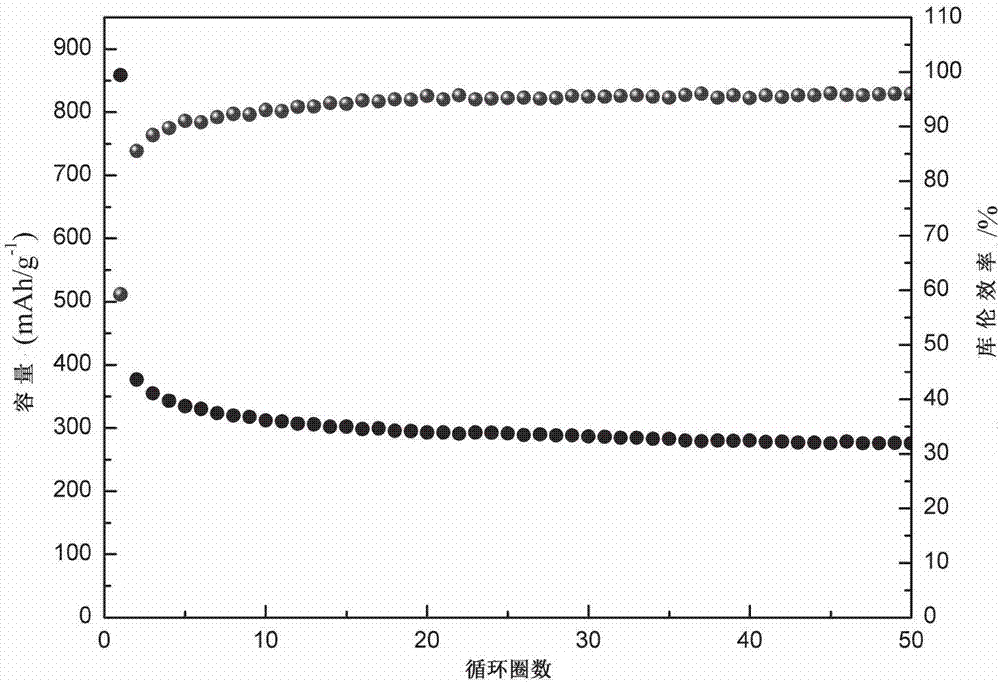
【图2】为实施例1制得的碳纳米片材料用于钠离子电池的循环圈数-循环放电容量、库伦效率图。
具体实施方式
以下实施例旨在对本发明内容做进一步详细说明;而本发明权利要求的保护范围不受实施例限制。
实施例1
将20ml1.0mol/l硝酸镍溶液、0.5ml聚乙二醇(分子量600,0.02mol)和0.5ml1mol/l的氢氧化钠搅拌均匀,进行水热反应,保温温度180℃,保温时间24h。然后采用去离子水洗涤3遍,然后采用无水乙醇洗涤2遍,然后放在真空干燥箱60℃干燥10h,得到金属纳米片。将0.08g金属纳米片(0.001mol)置于200ml溶有8.8g多巴胺(0.05mol),6.63g赖氨酸(0.045mol)的tris缓冲液中,调节ph值为8.5,静置12h,经过抽滤洗涤干燥,得到碳氮-金属纳米片前驱体。氮气气氛下将前驱体进行碳化,碳化温度1000℃,碳化时间为2h,升温速率为2℃/min。随后高温碳化产物采用稀酸溶液与水反复洗涤后,置于50℃温度条件下,真空干燥8h。得到较高氮掺杂量和碳层间距较大的碳纳米片材料。所述材料氮元素原子掺杂百分比为29%(原子个数百分含量),比表面积为782m2/g,碳层厚度33nm,层间距0.471nm,制得碳纳米片材料的扫描电镜图(sem)见图1。
采用本实施例制备的碳纳米片材料,加入10wt.%superp作为导电剂,10wt.%羧甲基纤维素钠(cmc)作为粘结剂,经研磨充分之后加入少量去离子水混合形成均匀的黑色糊状浆料,将这些浆料涂覆在铜箔集流体上作为测试电极,以金属钠片作为对比电极组装成为扣式电池,其采用电解液体系为1mnaclo4/ec:dec(1∶1),在50ma/g的电流密度下,测试循环性能;在1000ma/g、2000ma/g等不同的电流密度下测试电池的倍率性能。测试结果表明,本例制备的钠离子电池负极具有良好的电化学性能:在50ma/g的电流密度下,其首圈库伦效率为71%,首圈放电容量为366ma/g,循环100圈后,仍能保持291ma/g的比容量;在1000ma/g和2000ma/g的放电密度下,仍能分别保持241mah/g和201mah/g的比容量。制得半电池前50圈循环放电容量与库伦效率图见图2。
实施例2
将20ml1.0mol/l硝酸铜溶液、0.5ml聚乙二醇(分子量600,0.02mol)和0.5ml1mol/l的氢氧化钠搅拌均匀,进行水热反应,保温温度160℃,保温时间24h。然后采用去离子水洗涤3遍,然后采用无水乙醇洗涤2遍,然后放在真空干燥箱60℃干燥10h,得到金属纳米片。将0.8g金属纳米(0.01mol)片置于200ml溶有8.8g多巴胺(0.05mol),5.17g天冬酰胺(0.04mol)的tris缓冲液中,调节ph值为8.5,静置16h,经过抽滤洗涤干燥,得到碳氮-金属纳米片前驱体。氮气气氛下将前驱体进行碳化,碳化温度1000℃,碳化时间为2h,升温速率为2℃/min。随后高温碳化产物采用稀酸溶液与水反复洗涤后,置于50℃温度条件下,真空干燥8h。得到较高氮掺杂量和碳层间距较大的碳纳米片材料。所述材料氮元素原子掺杂百分比为22%(原子个数百分含量),比表面积为593m2/g,碳层厚度42nm,层间距0.403nm,采用本实施例制备的碳纳米片材料,加入10wt.%superp作为导电剂,10wt.%羧甲基纤维素钠(cmc)作为粘结剂,经研磨充分之后加入少量去离子水混合形成均匀的黑色糊状浆料,将这些浆料涂覆在铜箔集流体上作为测试电极,以金属钠片作为对比电极组装成为扣式电池,其采用电解液体系为1mnaclo4/ec:dec(1∶1),在50ma/g的电流密度下,测试循环性能;在1000ma/g、2000ma/g等不同的电流密度下测试电池的倍率性能。测试结果表明,在50ma/g的电流密度下,其首圈库伦效率为69%,首圈放电容量为354ma/g,循环100圈后,仍能保持277ma/g的比容量;在1000ma/g和2000ma/g的放电密度下,仍能分别保持233mah/g和188mah/g的比容量。
实施例3
将20ml1.0mol/l硝酸铁溶液、0.5ml聚乙二醇(分子量600,0.02mol)和0.5ml1mol/l的氢氧化钠搅拌均匀,进行水热反应,保温温度160℃,保温时间16h。然后采用去离子水洗涤3遍,然后采用无水乙醇洗涤2遍,然后放在真空干燥箱60℃干燥10h,得到金属纳米片。将0.08g金属纳米片(0.005mol)置于200ml溶有8.8g多巴胺(0.05mol),2.09g天冬氨酸的(0.02mol)tris缓冲液中,调节ph值为8.5,静置16h,经过抽滤洗涤干燥,得到碳氮-金属纳米片前驱体。氮气气氛下将前驱体进行碳化,碳化温度900℃,碳化时间为2h,升温速率为2℃/min。随后高温碳化产物采用稀酸溶液与水反复洗涤后,置于50℃温度条件下,真空干燥8h。得到较高氮掺杂量和碳层间距较大的碳纳米片材料。所述材料氮元素原子掺杂百分比为21%(原子个数百分含量),比表面积为446m2/g,碳层厚度38nm,层间距0.393nm,采用本实施例制备的碳纳米片材料,加入10wt.%superp作为导电剂,10wt.%羧甲基纤维素钠(cmc)作为粘结剂,经研磨充分之后加入少量去离子水混合形成均匀的黑色糊状浆料,将这些浆料涂覆在铜箔集流体上作为测试电极,以金属钠片作为对比电极组装成为扣式电池,其采用电解液体系为1mnaclo4/ec:dec(1∶1),在50ma/g的电流密度下,测试循环性能;在1000ma/g、2000ma/g等不同的电流密度下测试电池的倍率性能。测试结果表明,在50ma/g的电流密度下,其首圈库伦效率为70%,首圈放电容量为349ma/g,循环100圈后,仍能保持270ma/g的比容量;在1000ma/g和2000ma/g的放电密度下,仍能分别保持228mah/g和178mah/g的比容量。该实施例碳化温度较低,结晶效果偏差,致使其材料容量变低,倍率性能变差。
实施例4
将20ml1.0mol/l氯化钴溶液、0.5ml聚乙二醇(分子量600,0.02mol)和0.5ml1mol/l的氢氧化钠搅拌均匀,进行水热反应,保温温度150℃,保温时间20h。然后采用去离子水洗涤3遍,然后采用无水乙醇洗涤2遍,然后放在真空干燥箱60℃干燥10h,得到金属纳米片。将0.16g金属纳米片(0.05mol)置于200ml溶有8.8g多巴胺(0.05mol),2.96g赖氨酸(0.01mol)的tris缓冲液中,调节ph值为8.5,静置16h,经过抽滤洗涤干燥,得到碳氮-金属纳米片前驱体。氮气气氛下将前驱体进行碳化,碳化温度1000℃,碳化时间为2h,升温速率为2℃/min。随后高温碳化产物采用稀酸溶液与水反复洗涤后,置于50℃温度条件下,真空干燥8h。得到具有氮掺杂并碳层间距较大的碳纳米片材料。所述材料氮元素原子掺杂百分比为12%(原子个数百分含量),比表面积为381m2/g,碳层厚度31nm,层间距0.371nm,采用本实施例制备的碳纳米片材料,加入10wt.%superp作为导电剂,10wt.%羧甲基纤维素钠(cmc)作为粘结剂,经研磨充分之后加入少量去离子水混合形成均匀的黑色糊状浆料,将这些浆料涂覆在铜箔集流体上作为测试电极,以金属钠片作为对比电极组装成为扣式电池,其采用电解液体系为1mnaclo4/ec:dec(1∶1),在50ma/g的电流密度下,测试循环性能;在1000ma/g、2000ma/g等不同的电流密度下测试电池的倍率性能。测试结果表明,在50ma/g的电流密度下,其首圈库伦效率为75%,首圈放电容量为315ma/g,循环100圈后,仍能保持255ma/g的比容量;在1000ma/g和2000ma/g的放电密度下,仍能分别保持219mah/g和169mah/g的比容量。相较于其他实施例,本实施例氮源选择为含氮量低的赖氨酸,氮源用量少于优选范围,最终制得材料含氮量低,层间距相对较小,材料表面缺陷不丰富,活性位点较少,导致材料容量较低,倍率性能较差。
实施例5
和实施例1相比,区别仅在于,热处理(碳化)的温度为1200℃,所得材料氮元素原子掺杂百分比为28%(原子个数百分含量),比表面积为663m2/g,碳层厚度30nm,层间距0.412nm,;使用该材料组装为扣式电池,在相同条件下测试其电化学性能,在50ma/g的电流密度下,其首圈库伦效率为66%,首圈放电容量为301ma/g,循环100圈后,仍能保持266ma/g的比容量;在1000ma/g和2000ma/g的放电密度下,仍能分别保持210mah/g和199mah/g的比容量。碳化温度过高使碳纳米片材料无定型程度下降,晶型向石墨材料转变,导致容量降低。
对比例1
将20ml1.0mol/l硝酸镍溶液、0.5ml聚乙二醇(分子量600,0.02mol)和0.5ml的lmol/l的氢氧化钠搅拌均匀,进行水热反应,保温温度160℃,保温时间20h。然后采用去离子水洗涤3遍,然后采用无水乙醇洗涤2遍,然后放在真空干燥箱60℃干燥10h,得到金属纳米片。将0.16g金属纳米片(0.002mol)置于200ml溶有8.8g多巴胺(0.05mol)的tris缓冲液中,调节ph值为8.5,静置16h,经过抽滤洗涤干燥,得到碳氮-金属纳米片前驱体。氮气气氛下将前驱体进行碳化,碳化温度800℃,碳化时间为2h,升温速率为2℃/min。随后高温碳化产物采用稀酸溶液与水反复洗涤后,置于50℃温度条件下,真空干燥8h。得到较高氮掺杂量和碳层间距较大的碳纳米片材料。所述材料氮元素原子掺杂百分比为7%(原子个数百分含量),比表面积为233m2/g,碳层厚度55nm,层间距0.345nm,采用本实施例制备的碳纳米片材料,加入10wt.%superp作为导电剂,10wt.%羧甲基纤维素钠(cmc)作为粘结剂,经研磨充分之后加入少量去离子水混合形成均匀的黑色糊状浆料,将这些浆料涂覆在铜箔集流体上作为测试电极,以金属钠片作为对比电极组装成为扣式电池,其采用电解液体系为1mnaclo4/ec:dec(1∶1),在50ma/g的电流密度下,测试循环性能;在1000ma/g、2000ma/g等不同的电流密度下测试电池的倍率性能。测试结果表明在50ma/g的电流密度下,其首圈库伦效率为71%,首圈放电容量为311ma/g,循环100圈后,保持有249ma/g的比容量;在1000ma/g和2000ma/g的放电密度下,仍能分别保持188mah/g和145mah/g的比容量。和实施例1相比,对比例未添加氨基酸作为氮源,没有氮作为杂原子掺入材料,碳纳米片材料层间距变小,材料比容量下降,特别是长期稳定性下降明显。
对比例2
将20ml1.0mol/l硝酸镍溶液、0.5ml聚乙二醇(分子量600,0.02mol)和0.5ml1mol/l的氢氧化钠搅拌均匀,进行水热反应,保温温度130℃,保温时间24h。然后采用去离子水洗涤3遍,然后采用无水乙醇洗涤2遍,然后放在真空干燥箱60℃干燥10h,得到金属纳米片。将0.08g金属纳米片(0.001mol)置于200ml溶有8.8g多巴胺(0.05mol),6.63g赖氨酸(0.045mol)的tris缓冲液中,调节ph值为8.5,静置12h,经过抽滤洗涤干燥,得到碳氮-金属纳米片前驱体。氮气气氛下将前驱体进行碳化,碳化温度1000℃,碳化时间为2h,升温速率为2℃/min。随后高温碳化产物采用稀酸溶液与水反复洗涤后,置于50℃温度条件下,真空干燥8h。得到较高氮掺杂量和碳层间距较大的碳纳米片材料。所述材料氮元素原子掺杂百分比为29%(原子个数百分含量),比表面积为290m2/g,碳层厚度33nm,层间距0.451nm。采用制备的碳纳米片材料,加入10wt.%superp作为导电剂,10wt.%羧甲基纤维素钠(cmc)作为粘结剂,经研磨充分之后加入少量去离子水混合形成均匀的黑色糊状浆料,将这些浆料涂覆在铜箔集流体上作为测试电极,以金属钠片作为对比电极组装成为扣式电池,其采用电解液体系为1mnaclo4/ec:dec(1∶1),在50ma/g的电流密度下,测试循环性能;在1000ma/g、2000ma/g等不同的电流密度下测试电池的倍率性能。测试结果表明:在50ma/g的电流密度下,其首圈库伦效率为71%,首圈放电容量为303ma/g,循环100圈后,仍能保持261ma/g的比容量;在1000ma/g和2000ma/g的放电密度下,仍能分别保持201mah/g和155mah/g的比容量。和实施例1相比,对比例降低了水热反应温度,导致金属氧化物纳米片结构难以形成,最终导致碳纳米片材料结构团聚严重,倍率性能变差。
对比例3
本对比例探讨碳化采用较低的碳化温度,步骤如下:
将20ml1.0mol/l硝酸镍溶液、0.5ml聚乙二醇(分子量600,0.02mol)和0.5ml1mol/l的氢氧化钠搅拌均匀,进行水热反应,保温温度140℃,保温时间20h。然后采用去离子水洗涤3遍,然后采用无水乙醇洗涤2遍,然后放在真空干燥箱60℃干燥10h,得到金属纳米片。将0.16g金属纳米片(0.002mol)置于200ml溶有8.8g多巴胺(0.05mol),9.30g(0.01mol)甘氨酸的tris缓冲液中,调节ph值为8.5,静置16h,经过抽滤洗涤干燥,得到碳氮-金属纳米片前驱体。氮气气氛下将前驱体进行碳化,碳化温度800℃,碳化时间为2h,升温速率为2℃/min。随后高温碳化产物采用稀酸溶液与水反复洗涤后,置于50℃温度条件下,真空干燥8h。得到较高氮掺杂量和碳层间距较大的碳纳米片材料。所述材料氮元素原子掺杂百分比为12%(原子个数百分含量),比表面积为622m2/g,碳层厚度42nm,层间距0.371nm,采用本实施例制备的碳纳米片材料,加入10wt.%superp作为导电剂,10wt.%羧甲基纤维素钠(cmc)作为粘结剂,经研磨充分之后加入少量去离子水混合形成均匀的黑色糊状浆料,将这些浆料涂覆在铜箔集流体上作为测试电极,以金属钠片作为对比电极组装成为扣式电池,其采用电解液体系为1mnaclo4/ec:dec(1∶1),在50ma/g的电流密度下,测试循环性能;在1000ma/g、2000ma/g等不同的电流密度下测试电池的倍率性能。测试结果表明,在50ma/g的电流密度下,其首圈库伦效率为71%,首圈放电容量为311ma/g,循环100圈后,仍能保持249ma/g的比容量;在1000ma/g和2000ma/g的放电密度下,仍能分别保持200mah/g和150mah/g的比容量。低的碳化温度导致材料碳含量低,结晶效果差,最终导致材料容量变低,倍率性能较差。
- 还没有人留言评论。精彩留言会获得点赞!