氟取代的阳离子无序锂金属氧化物及其制备方法与流程
本发明涉及用于大容量锂离子电池电极的氟取代阳离子无序锂金属氧化物及其制备方法。
背景技术:
由于具有较高的能量和高功率性能,锂离子(“li-ion”)电池是目前研究最多的储能装置之一。随着对高性能锂离子电池需求的不断增加,人们从不同的化学空间中寻找高能量密度的阴极材料。尤其是氧化物材料,由于其在所有阴极材料中具有最高的能量密度而受到了人们的广泛关注。
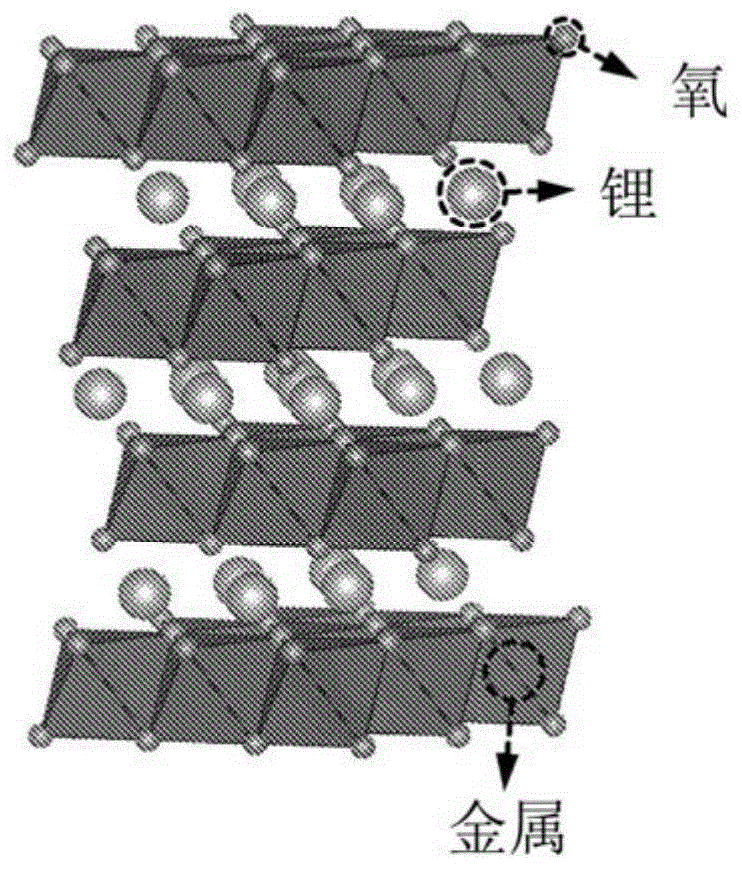
更具体地,层状锂过渡金属氧化物,如licoo2,已经成为可充电锂电池最重要的阴极材料之一。在这些材料中,锂离子和过渡金属离子被很好地分离,以形成在其晶体结构中交替的不同的层。在这些有序化合物中,从过渡金属亚晶格中分离锂(li)位置和路径(层状氧化物中的2d板),这提供了稳定性和电子存储能力。图1提供了层状岩盐结构li-m-o的示意图。在锂和过渡金属亚晶格之间几乎没有或没有混合的情况下,通常认为具有良好有序的结构对于获得具有良好循环寿命的高容量阴极材料是重要的。事实上,人们认为其结构中良好的层状性是材料中高的锂迁移率的必要条件,并且观察到阳离子的混合通过减缓锂的扩散而导致循环性差。这些观察结果可能导致研究人员忽视阳离子无序的锂过渡金属氧化物作为有前途的阴极材料。
近年来,对氧化物空间取得了重要的进展,拓宽了高能密度阴极材料的研究空间。具体地说,由于无序结构限制了锂的扩散,阳离子无序锂过渡金属氧化物(“li-tmoxides”)通常被认为是电化学惰性的,如果有足够的锂过量(即锂位点的数量多于tm位点的数量(li1+xtm1-xo2中x>0.09)),阳离子无序锂过渡金属氧化物(“li-tm氧化物”)可以作为有前途的阴极材料。实际上,一旦引入足够的过量锂,就可以在无序结构中进行简单的锂扩散,这反过来又引入了易于扩散的锂扩散通道(0-tm通道)的渗透网络,通过此网络,由于缺少排斥性tm离子,活化的li+离子在扩散时的弱静电斥力使得锂的扩散在无序结构中是容易的。
然而,在提供高能量密度阴极材料方面,阳离子无序/阳离子混合仍然存在许多困难和挑战。例如,氧气氧化(通常是从无序材料中获得高容量所必需的)可以通过晶格致密化引发氧气损失,这会通过降低锂过量水平(特别是在表面附近的)来降低无序材料中的0-tm渗流(从而降低锂扩散)。因此,几乎所有阳离子无序的li-tm氧化物,其tm氧化还原剂(例如,fe2+/4+、ni2+/4+、co2+/4+)与氧气氧化还原剂重叠,氧损失后出现较大的极化,显示出有限的循环性。此外,氧损失也会导致如li2co3层的电阻表面层,这会进一步增加阴极的阻抗。
kang等人的美国专利no.7,205,072中描述了一种先前报道的用于改善层状(岩盐)材料稳定性的策略。kang描述了在层状锂镍-锰-钴基氧化物材料上的氟替代策略,其改善了层状(岩盐)材料的抗无序、结构稳定性。层状岩盐与阳离子无序岩盐之间的结构选择很大程度上取决于材料的组成。在kang中讨论的氟取代的锂镍-锰-钴氧化物,基于组合物中包含的元素的组成构成和氧化态,完全形成层状岩盐结构。kang没有讨论氟取代对阳离子无序结构的结构完整性的作用。
因此,仍然需要具有改进的电化学性能的阳离子无序锂过渡金属氧化物以用作阴极材料。
技术实现要素:
本发明涉及一种锂金属氧化物,其通式为:li1+xm1-xo2-yfy,所述锂金属氧化物具有阳离子无序的岩盐结构,其中0.05≤x≤0.3,0<y≤0.3且m是过渡金属。在一个实施例中,通式可包括0.09≤x≤0.3和0.10≤y≤0.25。在另一实施例中,m选自ti、v、cr、mn、fe、co、ni、zr、nb、mo、sn、sb及其组合。例如,m可以选自mo、ni、ti及其组合。在又一实施例中,本发明的锂金属氧化物可具有通式:li1+xniatibmo1-x-a-bo2-yfy,其中0.2≤a≤0.6且0.2≤b≤0.6。
本发明的锂金属氧化物可具有阳离子无序的岩盐结构,其特征在于晶体空间群
本发明还涉及一种制造具有通式li1+xm1-xo2-yfy的锂金属氧化物的方法,所述锂金属氧化物具有阳离子无序的岩盐结构,其中0.05≤x≤0.3且0<y≤0.3,且m为过渡金属,例如ti、v、cr、mn、fe、co、ni、zr、nb、mo、sn、sb或其组合,该方法包括以下步骤:提供锂基前体、过渡金属(m)基前体和氟基前体;将所述锂基前体、过渡金属基前体和氟基前体分散在有机溶剂中,以获得前体悬浮液;优选地,研磨所述前体悬浮液,使得平均粒径为约100nm至约180nm;将所述前体悬浮液干燥并造粒;在氧气的存在下,在至少600℃的温度下烧制所述前体悬浮液。
在一个实施例中,将化学计量量的所述锂基前体、过渡金属基前体和氟基前体分散到溶剂中。锂基前体可以是li2co3,氟基前体可以是lif。在另一实施例中,有机溶剂选自丙酮或乙醇。在另一个实施例中,研磨步骤可选自球磨、振动研磨和高能球磨。前体悬浮液可在氧气存在下在至少700℃的温度下焙烧约10小时。
本发明还涉及包含根据本发明的锂金属氧化物的正极材料。本发明还涉及根据本发明的锂离子电池,包括负极材料、电解液和包括锂金属氧化物的正极材料。本发明的锂离子电池可用于便携式电子设备、汽车或储能系统。
附图说明
根据结合下面描述的附图提供的以下详细描述,可以确定本发明的进一步特征和优点。
图1是层状岩盐结构的示意图;
图2是根据本发明的一个实施例的无序-岩盐结构的示意图;
图3示出了可充电锂离子电池的示意图;
图4a示出了li1.15ni0.45ti0.3mo0.1o1.85f0.15(“本发明的lnf15”);li1.2ni0.333ti0.333mo0.133o2(“对比的ln15”)和li1.2ni0.333ti0.333mo0.133o2(“对比的ln20”)的x射线衍射(“xrd”)图和细化结果;
图4b示出了本发明的lnf15粒子上的能量色散光谱(“eds”)映射;
图5a-5d示出了在室温下以20ma/g的速度在1.5-4.6v之间循环时,对比的ln15、对比的ln20、本发明的lnf15和本发明的hb-lnf15的初始五个周期电压分布;
图6a示出了对比的ln15、对比的ln20、本发明的lnf15和本发明的hb-lnf15的第一周期和第二充电电压曲线;
图6b示出对比的ln15、对比的ln20、本发明的lnf15和本发明的hb-lnf15的第一周期电压曲线;
图6c示出了对比的ln20和本发明的lnf15在第一次充电至270mah/g后通过恒电流间歇滴定试验(“gitt”)得到的第一放电电压曲线;
图7a-7d分别示出了对比的ln15、对比的ln20、本发明的lnf15和本发明的hb-lnf15当每个化合物在室温下以20ma/g的速度充电和以10ma/g、20ma/g、40ma/g、100ma/g和200ma/g的速度在1.5v和4.6v之间放电时的放电电压曲线;
图8a-8c分别示出了对比的ln15、对比的ln20和本发明的lnf15在充电至4.8v然后以20ma/g放电至1.5v时的电压分布,以及氧和二氧化碳的差分电化学质谱分析结果;
图9a示出了对比的ln20的原位xrd图;
图9b示出来自对比的ln20的rietveld精修(rietveldrefinements)后的电压分布和晶格参数;
图9c示出了本发明的lnf15的原位xrd图;
图9d示出了来自本发明的lnf15的rietveld精修的电压分布和晶格参数;
图10a示出了使用“体积敏感的”总荧光产额(“tfy”)模式在本发明的hb-lnf15的镍的l边缘上的软x射线吸收光谱(“sxas”);
图10b示出了使用“表面敏感的”总电子产额(“tey”)模式的本发明hb-lnf15的镍的l边缘上的sxas;
图10c示出使用tfy模式在本发明hb-lnf15的氧的k边缘上的sxas;
图10d示出使用tey在本发明hb-lnf15的氧的k边缘上的sxas测试;
图11a示出li1.25ni0.35ti0.3mo0.1o1.75f0.25(“本发明的l125ntmof”)的xrd图谱;
图11b示出了本发明的l125ntmof在室温下以20ma/g在1.5和4.5v之间循环时的电压曲线;
图12a示出li1.2ni0.5nb0.3o1.7f0.3(“本发明的l120nnof”)的xrd图;以及
图12b示出了本发明的l120nnof在室温下以20ma/g在1.5和4.5v之间循环时的电压曲线。
具体实施方式
本发明涉及氟取代的阳离子无序的锂金属氧化物(“li-m氧化物”)。在不受任何特定理论约束的情况下,人们认为,在阳离子无序的锂金属氧化物材料中,氟取代氧可以通过减少循环过程中的氧损失来改善材料的电化学性能。众所周知,氧损失通过触发阳离子致密化降低li-m氧化物材料的电化学性能,阳离子致密化降低锂过量程度(xinli1+xtm1-xo2-yfy),这是无序锂过量材料中使得锂扩散容易所必需的。
人们认为,氟化可以通过提高无序锂过量材料中氧化还原活性过渡金属的含量来改善过渡金属的氧化还原,从而防止高脱锂时氧氧化还原过多,从而减少氧损失。换言之,氟取代抑制了氧损失,反过来改善了锂过量无序阴极材料的循环性能。事实上,通过氟取代来减轻无序锂过量化合物的氧损失,通过减少充放电时的极化,显著改善了循环性能。因此,本发明的氟取代材料具有高容量和高电压,这是其他类型的镍氧化还原基(或钴、铁氧化还原基)阳离子无序阴极材料所无法实现的。
本发明提供氟取代的阳离子无序锂金属氧化物(“li-m氧化物”)。在一个实施例中,本发明的li-m氧化物包括阳离子无序岩盐结构。实际上,本文所讨论的li-m氧化物仅形成无序岩盐结构。如本文所用,阳离子无序岩盐结构是指以晶体空间群
本发明的阳离子无序岩盐结构提供了氧取代氟的方法。如上所述,所取代的氟氧的fcc框架相同,并随氧随机分布。实际上,本发明的无序li-m氧化物是阳离子完全混合的,即100%的阳离子混合。图2示出本发明设想的阳离子无序岩盐结构。如图2所示,在岩盐结构中,氧/氟、锂和过渡金属的分布是完全胡乱的。
在一个实施例中,本发明的li-m氧化物可形成层状岩盐,并在充放电循环时转变为无序岩盐。在另一实施例中,本发明的li-m氧化物最初形成为无序岩盐材料。然而,本发明考虑使用阳离子无序li-m氧化物作为最终成分。
在一个实施例中,本发明的li-m氧化物具有通式(1):
li1+xm1-xo2-yfy(1)
其中0.05≤x≤0.3且0<y≤0.3。在这方面,通式(1)可定义为0.09≤x≤0.3且0.10≤y≤0.25。在另一方面,通式(1)可定义为0.1≤x≤0.25且0.10≤y≤0.15。根据本发明,通式(1)的m可以是过渡金属。在一个实施例中,可从ti、v、cr、mn、fe、co、ni、zr、nb、mo、sn、sb或其组合中选择m。在另一实施例中,可从mo、ni、ti或其组合中选择m。
在这方面,通式(1)中li与m之比可在1.05:0.95至1.3:0.7之间。例如,li与m之比可以在1.1:0.99到1.2:0.8之间。在另一实施例中,通式(1)中的o与f之比可在1.99:0.01至1.7:0.3之间。例如,o与f之比可能在1.90:0.1到1.75:0.25之间。
在另一实施例中,本发明的li-m氧化物具有通式(2):
li1+xm1-x-ym’yo2-zfz(2)
其中0.05≤x≤0.3,0<y≤0.8,0<z≤0.3。在另一实施例中,通式(2)可定义为0.05≤x≤0.3,0<y≤0.7,0<z≤0.3。在这方面,定义了通式(2)的x和y的值,使得m大于或等于0。根据本发明,通式(2)的m可以是从v、cr、mn、fe、co、ni、mo或其组合中选择的过渡金属。在另一实施例中,m’可以是从ti、mo、nb、sb、zr或其组合中选择的过渡金属。在这方面,通式(2)的m和m'由不同的过渡金属定义。
在另一实施例中,本发明的li-m氧化物可包括锂镍钛钼氧氟化物。例如,本发明的li-m氧化物可具有通式(3):
li1+xniatibmo1-x-a-bo2-yfy(3)
其中0.05<x<0.3,0.2≤a≤0.6,0.2≤b≤0.6,0<y≤0.3。在这方面,通式(3)的li-m氧化物可包括化合物li1.15ni0.45ti0.3mo0.1o1.85f0.15,其中x=0.15,a=0.45,b=0.3,y=0.15。通式(3)的li-m氧化物还可包括化合物li1.25ni0.35ti0.3mo0.1o1.75f0.25,其中x=0.25,a=0.35,b=0.3,y=0.25。
在另一个实施例中,本发明的li-m氧化物可包括锂镍铌氧氟化物。例如,本发明的li-m氧化物可具有通式(4):
li1+xnianbbo2-yfy(4)
其中0.05<x<0.3,0.2≤a≤0.6,0.2≤b≤0.6,0<y≤0.3。例如,通式(4)的li-m氧化物可包括化合物li1.2ni0.5nb0.3o1.7f0.3,其中x=0.2,a=0.5,b=0.3,y=0.3。
本发明的li-m氧化物可具有不同的平均粒子大小。在一个实施例中,本发明的li-m氧化物具有约10nm到约10μm的平均(初级)粒子大小。在另一实施例中,本发明的li-m氧化物具有约100nm至约200nm的平均(初级)粒子大小。在另一实施例中,本发明的li-m氧化物具有约100nm至约180nm的平均(初级)粒子大小。
本发明的li-m氧化物还可以具有可变的晶格参数。例如,本发明的li-m氧化物可具有约
本发明还包括制造本发明的氟取代的阳离子无序锂金属氧化物(“li-m氧化物”)的方法。各种方法可用于制备本发明的li-m氧化物,包括但不限于固态反应方法、水溶液方法或机械化学合成方法。在一个实施例中,固态反应方法可用于制备本发明的li-m氧化物。在这方面,本发明的li-m氧化物的制造方法包括提供用于制造li-m氧化物的必要前体的步骤。例如,该过程可包括提供至少一种锂基前体、至少一种过渡金属前体和至少一种氟基前体的步骤。对于本领域普通技术人员显而易见的是,提供所需氟取代阳离子无序li-m氧化物的元素成分的任何前体均可在本发明中使用。然而,在一个实施例中,锂基前体可包括li2co3、li2o或lioh。类似地,优选的氟基前体包括lif。
在选择所需的前体之后,可将化学计量量的锂基前体、过渡金属基前体和氟基前体分散在溶剂中以获得前体悬浮液。在一个实施例中,溶剂可包括任何包括极性或非质子性溶剂的有机溶剂。本发明涉及的适当溶剂包括但不限于丙酮、乙酸、乙腈、苯、丁醇、四氯化碳、二甘醇、二乙醚、乙醇、乙酸乙酯、乙二醇、异丙醇、甲醇、戊烷、丙醇、甲苯和二甲苯氖。在一个实施例中,所使用的溶剂可以是乙醇。在另一实施例中,所使用的溶剂可以是丙酮。
在形成前体悬浮液后,随后可以对所得悬浮液进行研磨。在一个实施例中,对所得到的悬浮液可以进行球磨。在另一个实施例中,对所得到的悬浮液可进行振动筛研磨。在另一个实施例中,所得悬浮液可经高能球磨以降低化合物的平均初级粒子大小。前体悬浮液可经历约1小时至约50小时的研磨。在另一实施例中,前体悬浮液可经研磨约10小时至约20小时。例如,前体悬浮液可经历约15小时的研磨。
在本实施例中,在研磨完成后,可在烘箱中干燥前体悬浮液。可以将前体悬浮液干燥约3小时至约50小时。在另一实施例中,可将前体悬浮液干燥约10小时至约20小时。干燥后,可以将前体悬浮液造粒并在氧气存在下在约400℃至约1200℃的温度下烧制。在另一个实施例中,可在约600℃至约1200℃的温度下烧制颗粒。在另一个实施例中,可在约700℃至约1200℃的温度下烧制颗粒。烧制的持续时间可根据使用的温度而变化。在一个实施例中,可将颗粒烧制约30分钟至约40小时。在另一个实施例中,可将颗粒烧制约5小时至约15小时。例如,可在700℃的温度下在氧气存在下将该颗粒燃烧10小时。烧成后,手动将颗粒磨成细粉。
本文所描述的氟取代的阳离子无序li-m氧化物提供了改进的电化学性能。在本发明的阳离子无序li-m氧化物中提供的氟取代导致降低氧损失和极化,从而导致循环性能的改善。例如,与非氟化li-m氧化物相比,在氧损失降低的情况下,本发明的氟化li-m氧化物在循环时的电压极化显著降低。事实上,本发明的氟化li-m氧化物在充电和放电过程中表现出减小的电压间隙,表明氟化降低了极化。减小的极化可以提供高放电容量和增加的平均放电电压。在一个实施例中,本发明的氟化li-m氧化物具有至少约180mah/g的放电容量。在另一个实施例中,本发明的氟化li-m氧化物具有至少约210mah/g的放电容量。例如,本发明的氟化li-m氧化物具有约180mah/g到约330mah/g的放电容量。
此外,本发明的氟化li-m氧化物具有约2.3v到约3.8v的平均放电电压和约500wh/kg到约1000wh/kg的放电能量密度。在一个实施例中,本发明的氟化li-m氧化物具有至少约3.0v、优选3.25v的平均放电电压和高于750wh/kg的放电能量密度。本文所描述的氟化li-m氧化物显示出比非氟化li-m氧化物高出2.5v以上的放电容量。例如,本发明的氟化li-m氧化物在2.5v以上提供约180mah/g的放电容量。实际上,本发明的氟化li-m氧化物在3v以上的电压下提供高于150mah/g的放电容量。
此外,本发明的氟化li-m氧化物显示出改进的容量保持性。在一个实施例中,本发明的氟化li-m氧化物以20ma/g在1.5-4.6v之间循环20次后,显示出至少80%的初始放电容量的改进的容量保持性。在另一个实施例中,本发明的氟化li-m氧化物以20ma/g在1.5-4.6v之间循环20次后,显示出至少85%的初始放电容量的改进的容量保持性。这种改进的容量保持性至少比无氟无序li-m氧化物的容量保持性高3%左右。事实上,本发明的氟化li-m氧化物的容量保持性至少比非氟化无序li-m氧化物的容量保持性高5%左右。
在不受任何特定理论约束的情况下,相信本发明的氟化li-m氧化物相对于非氟化li-m氧化物的性能改进可归因于氧损失的减少。实际上,本发明的氟化li-m氧化物在比非氟化li-m氧化物更高的电压下经历氧损失。例如,本发明的氟化li-m氧化物中的氧气的演化延迟到至少4.4v(在第一次充电到4.8v时)。在另一个实施例中,本发明的氟化li-m氧化物中的氧气的演化延迟到至少4.5v(在第一次充电到4.8v时)。在另一个实施例中,本发明的氟化li-m氧化物中的氧气的演化延迟到至少4.6v(在第一次充电到4.8v时)。在本发明的这个方面,本发明的氟化li-m氧化物在比非氟化li-m氧化物高约0.15v的电压下经历氧损失。例如,本发明的氟化li-m氧化物在比非氟化li-m氧化物高约0.25v的电压下经历氧损失。
本发明还提供锂电池和锂离子电池,其包括电极材料,例如阴极,其由如本文所述的氟化阳离子无序li-m氧化物组成。在一个实施例中,根据本发明生产的氟取代的阳离子无序锂金属氧化物可以用作锂离子可再充充电电池中的阴极。图3显示了可再充电锂离子电池的示意图。如图3所示,锂离子在阴极10和阳极20之间的可逆穿梭使得可再充电锂离子电池30成为可能。本文所述的氟化阳离子无序li-m氧化物可作为锂离子可再充电电池中的阴极,用于如便携式电子设备的产品、包括电动汽车和混合动力汽车的汽车以及储能系统。
实例
以下非限制性实施例展示了根据本发明制备的氟取代的阳离子无序的锂金属氧化物。这些实施例仅用于说明本发明的优选实施方案,不应解释为本发明受到限制,本发明的范围由所附权利要求限定。
例1
实验程序
合成了如下氟取代的阳离子无序锂金属氧化物:
li-ni-ti-mo-o-f型,通式为:li1+xniatibmo1-x-a-bo2-yfy,0.05<x<0.3,0.2<a<0.6,0.2<b<0.6,0<y≤0.3,其中x=0.15,a=0.45,b=0.3,y=0.15(li1.15ni0.45ti0.3mo0.1o1.85f0.15)(“本发明的lnf15”)。
在制备本发明化合物时,将li2co3(alfaaesar,acs,99%分钟)、nico3(alfaaesar,99%)、tio2(alfaaesar,99.9%)、moo2(alfaaesar,99%)和lif(alfaaesar,99.99%)用作前体。将化学计量量的前体分散在丙酮中并球磨15小时,然后在烘箱中干燥过夜。将前体混合物造粒,然后在700℃下在空气中燃烧10小时,然后在炉中冷却至室温。烧成后,手动把颗粒磨成细粉。
还合成了以下对比的阳离子无序的金属锂氧化物(无氟取代):
li-ni-ti-mo-o型,通式为:li1+x/100ni1/2-x/120ti1/2-x/120mox/150o2,其中x=15(li1.15ni0.375ti0.375mo0.1o2)(“对比的ln15”);以及
li-ni-ti-mo-o型,通式为:li1+x/100ni1/2-x/120ti1/2-x/120mox/150o2,其中x=20(li1.2ni0.333ti0.333mo0.133o2)(“对比的ln20”)。
在制备比较化合物时,使用li2co3(alfaaesar,acs,99%分钟)、nico3(alfaaesar,99%)、tio2(alfaaesar,99.9%)、以及moo2(alfaaesar,99%)作为前体。将化学计量量的前体分散在丙酮中并球磨15小时,然后在烘箱中干燥过夜。将前体混合物造粒,然后在750℃下在空气中燃烧两小时,然后在炉中冷却至室温。烧成后,手动把颗粒磨成细粉。
为了制备阴极薄膜,将本发明的lnf15、对比的ln15和对比的ln20中每个的粉末分别与碳黑(timcal,superp)以70:20的重量比混合。将聚四氟乙烯(ptfe,dupont,teflon8c)(“ptfe”)作为粘合剂添加到各混合物中。每一个由此产生的阴极膜包括各自的重量比为70:20:10的本发明的lnf15、对比的ln15或对比的ln20以及碳黑和ptfe。将这些组分手动混合30分钟,并在充满氩气的手套箱内卷成薄膜。在一些情况下,使用高能球磨(retschpm200)将锂金属氧化物组分与碳黑以300rpm至500rpm的速率混合2至6小时。为了组装用于常规循环测试的电池,1m碳酸乙烯酯(“ec”)-碳酸二甲酯(“dmc”)溶液(1:1,technosemichem)的lipf6、玻璃微纤维过滤器(gewhatman)和锂金属箔(fmc)分别用作电解质、隔膜和对电极。将2032纽扣电池组装在充满氩气的手套箱内,并在室温下以恒电流模式在电池测试仪(arbin)上进行测试。阴极膜的负载密度约为5mg/cm2。比容量根据阴极膜中锂金属氧化物组分的量(即70%重量)计算。
在rigakuminiflex(cu源)上在5-85°的2θ范围内收集所制备的化合物的x射线衍射(“xrd”)图。在zeissgeminiultra-55analyticalfieldemissionsem上收集扫描电子显微镜(“sem”)图像。使用直流等离子体发射光谱法(astme1097-12)对li、ni、ti和mo进行化合物的元素分析。通过离子选择性电极(astmd1179-10)测定氟含量。
图4a描绘了本发明的lnf15、对比的ln15和对比的ln20化合物的xrd图案和精制结果。图4a以及下表1确认,通过标准固态反应成功制备了锂过量无序的氧化物岩盐相(无氟取代)和氟取代氧化物。如图4a所示,xrd精制显示晶格参数随着锂过量水平从15(ln15:
表格1:目标与测量锂:镍:钛:钼:比较ln15,比较ln20和本发明的lnf15的f原子比(其中,material:材料,target:目标;measured:测量的)
在图4a中的每个xrd图案的右侧的插图显示了每种材料的sem图像。如图4a所示,本发明的的lnf15(约180nm)的平均(初级)粒子大小大于对比的ln15(约100nm)和对比的ln20(约100nm)的平均(初级)粒子大小。为了进行比较,制备了与ln15和ln20具有类似粒子大小的额外的lnf15粉末。更具体地说,在lnf15上以300rpm的速度进行4小时的高能球磨,以将lnf15的平均初级粒子大小减小到100纳米。所得化合物在此称为“本发明的hb-lnf15”。
图4b显示了本发明的lnf15粒子上的能量色散光谱(“eds”)映射(ni、ti、o、mo、f)。根据eds图,在本发明的lnf15粒子中可以看到氟和其他元素的均匀分布。即,图4b进一步证实氟在无序晶格中被取代而不是形成第二相,并且成功地产生了目标lnf15相。
恒电流循环试验
为了比较本发明的lnf15、本发明的hb-lnf15、对比的ln15和对比的ln20化合物的电化学性质,对每种化合物进行恒电流循环试验。每种化合物在室温下以20ma/g在1.5-4.6v之间循环。
图5a-5d示出了在室温下以20ma/g在1.5-4.6v之间循环时,对比的ln15(图5a)、对比的ln20(图5b)、本发明的lnf15(图5c)和本发明的hb-lnf15(图5d)的初始五个周期电压分布。图5a-5d的插图显示材料在20个循环期间的容量保持性。换句话说,图5a-5d的插图显示了重复循环期间的充电和放电容量。
如图5a和5b所示,在20ma/g时,对比的ln15和对比的ln20分别能提供高达194mah/g(587wh/kg,2454wh/1)和220mah/g(672wh/kg,2775wh/1)的放电容量。然而,对比的ln15和对比的ln20的电压分布显示出充放电之间的大极化(电压间隙)。此外,大部分放电电压低于2.5v,导致对比的ln15和对比的ln20的平均放电电压分别约为3.03v和约3.05v。
相反,如图5c和图5d所示,本发明的lnf15和本发明的hb-lnf15能够以减小的极化进行循环,这使得能够分别提供210mah/g(681wh/kg,2894wh/1)和218mah/g(716wh/kg,3043wh/1)的高放电容量。随着极化的减小,本发明的lnf15和本发明的hb-lnf15的平均放电电压也增加到约3.25v。
极化试验
为了更直接地比较本发明的lnf15、本发明的hb-lnf15、对比的ln15和对比的ln20中的极化,图5a示出了每个化合物的第一周期和第二充电电压曲线。如图6a所示,在对比的ln15和对比的ln20的充放电中间可以看到很大的电压间隙。然而,在本发明的lnf15中减小了该电压间隙,并且在本发明的hb-lnf15中进一步减小,这表明氟化导致更少的极化,特别是在充电和放电中间。
图6b比较了本发明的lnf15、本发明的hb-lnf15、对比的ln15和对比的ln20化合物中的每一个的第一周期电压曲线。如图6b所示,比较本发明的hb-lnf15和对比的ln15,本发明的hb-lnf15提供在2.5v以上的80mah/g的更高放电容量,并且比对比的ln15提供高于3v的50mah/g的更高的放电容量。尽管如此,在1.5v(194mah/g至218mah/g)以上的放电容量几乎没有变化。
还进行了恒电流间歇滴定试验(“gitt”)以分析本发明的lnf15中极化减少的性质。在试验中,以20ma/g的速率,每一个步骤进行恒电流充电或放电10mah/g。每进行一个步骤就缓和五个小时。总的充放电容量分别为250mah/g。图6c显示了对比的ln20和本发明的lnf15在第一次充电到270mah/g后来自gitt的第一放电电曲线。图6c的插图示出了在约206小时内的电压-时间gitt曲线,其对应于电压-容量曲线中的大约110mah/g的放电容量。如图6c所示,电压-容量gitt曲线表明,与对比的ln20相比,在本发明lnf15中,尤其是在放电中间,每个放电步骤后的垂直位移(缓和)减小。图6c的插图中的电压-时间曲线显示,与对比的ln20相比,本发明的lnf15中的电压弛豫的时间相关部分小得多。这表明,在本发明的lnf15中,来自缓慢锂扩散的质量传输阻力减小,从而导致极化降低。
速率能力测试
进行速率性能测试以进一步研究本发明的lnf15、本发明的hb-lnf15、对比的ln15和对比的ln20化合物中的动力学。图7a-7d示出了对比的ln15(图7a)、对比的ln20(图7b)、本发明的lnf15(图7c)和本发明的hb-lnf15(图7d)当每种化合物以20ma/g的速度充电并在室温下以10ma/g、20ma/g、40ma/g、100ma/g和200ma/g的不同速率在1.5v到4.6v之间放电时的放电电压曲线。
从图7a-7d中可以看出,当放电速率从10ma/g增加到200ma/g时,对比的ln15的放电容量从206mah/g降低到151mah/g,对比的ln20的放电容量从234mah/g降低到180mah/g,本发明的lnf15的放电容量从214降低到148mah/g,本发明的hb-lnf15的放电容量从216降低到167mah/g。比较对比的ln15、对比的ln20和本发明hb-lnf15(均具有可比较的粒子大小)的速率能力,可以看出,尽管它们的速率能力差别不大,但相同速率中放电中间的极化减少导致本发明hb-lnf的能量密度高于比较ln15或比较ln20。例如,当以100ma/g放电时,本发明的hb-lnf递送的能量密度为592wh/kg,而对比的ln15递送的能量密度为493wh/kg,对比的ln20递送的能量密度为563wh/kg。
差分电化学质谱测量
对对比的ln15、对比的ln20和本发明的的lnf15进行了差示电化学质谱(dems)测量。在试验中,每种化合物在室温下以20ma/g的速度在1.5至4.8v之间循环。图8a-8c显示了对比的ln15(图8a)、对比的ln20(图8b)和本发明的的lnf15(图8c)在充电至4.8v、然后以20ma/g放电至1.5v时的电压曲线,以及氧和二氧化碳的差分电化学质谱分析结果。如图8a-8c所示,本发明的lnf15比对比的ln15和对比的ln20具有更少的氧损失。当第一次充电到4.8v时,对比的ln15和对比的ln20的氧气在大约4.35v(约185mah/g)后开始形成。也就是说,图8a和图8b示出了当充电高于约4.35v时对比的ln15和对比的ln20的氧损失。然而,对于本发明的lnf15,氧气的形成延迟至高于约4.5v(约220mah/g)。因此,与对比的ln15和对比的ln20相比,本发明的lnf15显示出更少的氧损失。
此外,对于对比的ln20和对比的ln15,第一次充电后的氧气释放总量也从0.26144μmol/mg和0.3975μmol/mg(每mg活性物质的气体物质的μmol)分别降低至本发明lnf15的0.07276μmol/mg。这些氧气释放量分别对应于对比的ln15、对比的ln20和本发明lnf15中总氧含量的2.341%、3.523%和0.7142%的损失。此外,氧气的形成发生在放电的最初阶段。因此,对于对比的ln15、对比的ln20和本发明的lnf15,氧气释放总量进一步增加到0.3010μmol/mg、0.4918μmol/mg和0.0915μmol/mg。
第一次充电时二氧化碳气体的释放也如图8a-8c所示。如图8a-8c所示,在所有情况下,二氧化碳气体在约4.4v以上形成。然而,在高于4.6v时,本发明的lnf15的二氧化碳气体释放速率(μmol/mg/min)远低于对比的ln15和对比的ln20。
原位xrd比较
为了研究氟取代对循环过程中结构变化的影响,对对比的ln20和本发明的lnf15进行了原位xrd分析。原位细胞在10ma/g的室温下在1.5–4.6v之间循环。对比的ln20和本发明的lnf15的原位xrd图分别在图9a和9c中显示。图9b和9d分别示出了对比的ln20和本发明的lnf15的电压曲线(由实心黑线表示)和来自rietveld精制(由黑色虚线表示)的晶格参数。
从图9a和图9c可以看出,对比的ln20和本发明的lnf15在充电时(002)峰值移向更高的角度,在放电时峰值移向更低的角度。这表明对比的ln20和本发明lnf15的晶格参数电荷在充电时减少并且在放电时增加。更具体地,对比的ln20的晶格参数从
如图9b和9d所示,在第一次充电的中间观察到对比的ln20和本发明lnf15的行为的最大差异。在对比的ln20充电超过约120mah/g后,晶格参数几乎没有降低,直到充电约215mah/g。然而,这种行为在本发明的lnf15中并不明显。
软x射线吸收光谱试验
为了研究本发明的hb-lnf15的氧化还原机制,对本发明的hb-lnf15进行了软x射线吸收光谱(“sxas”)试验,试验采用“体积敏感的”总荧光产额(tfy)模式和“表面感感的”总电子产额(tey)模式。如图10a-10d所示为镍的l边光谱和氧的k边光谱。图10a显示了使用tfy模式在本发明hb-lnf15的镍的l边缘上进行的sxas测试。图10b显示了使用tey模式在本发明的hb-lnf15的镍的l边缘上进行的sxas测试。图10c显示了使用tfy模式在本发明的hb-lnf15的氧的k边缘上的sxas测试。图10d显示了使用tey模式在本发明的hb-lnf15的氧的k边缘上的sxas测试。在这些试验中,在循环本发明的hb-lnf15之前、充电至70mah/g、140mah/g、210mah/g、4.6v(~280mah/g)之后和放电至1.5v之后收集数据。在室温下,以20mah/g循环本发明hb-lnf15。
图10a和10b分别显示了使用tfy模式和tey模式的本发明hb-lnf15的镍的l边缘光谱。金属的l边缘是由金属2p电子激发到未填充的三维轨道引起的,它可以分为两个区域:低光子能量的l3边缘和高能量的l2边缘,这是由于2p核孔自旋轨道分裂引起的。
从图10a所示的tfy模式(其是体积敏感的)的镍的l边缘光谱可以看出,循环前镍的l3边缘光谱在约853ev处具有最强的峰值,在约855ev处具有较小的峰值。当充电到70mah/g、140mah/g、210mah/g和4.6v(约280mah/g)时,两个峰值之间的强度比[=i(853ev)/i(855ev)]减小,并在第一次放电到1.5v(约225mah/g)后恢复到原始lnf15的比率。此外,在873ev附近,镍的l2边缘的光谱重量随充电而增大,随放电而减小。这表明,本发明的hb-lnf15化合物中的大部分ni2+氧化为ni3+和ni4+,并在放电后还原为ni2+。尽管如此,即使充电到4.6v,镍的l边缘的变化也很小,ni2+信号仍然很强。这表明,在循环过程中,在本发明hb-lnf15中仅使用了少量的ni2+/ni4+氧化还原。
从图10b所示的tey模式(其是表面敏感的(~10nm))的镍的l边缘光谱可以看出,镍边缘的变化甚至小于tfy模式下的镍边缘的变化。这表明,在循环过程中,表面区域的大部分镍离子以ni2+的形式存在,在氧化还原过程中的参与程度很小。
从本发明的hb-lnf15(图10c和图10d)的氧的k边缘光谱可以看出,大部分的ni2+在充电时被氧化,但表面的镍几乎没有被氧化。氧的k边缘是由氧的1s电子激发到未填充的氧的2p态引起的。
图10c显示了来自tfy模式的氧的k边缘光谱。如图10c所示,大约528.5ev的峰值在充电至4.6v时增大,在放电时消失。在大约528.5ev处的峰值生长可归因于大部分使用镍控制的(例如电子)的部分ni2+/ni4+氧化,这部分导致了由于(例如状态)共价性质而导致的空穴掺杂到氧的2p态。大部分这种镍的氧化与从tfy模式观察到的镍的l边缘光谱中的轻微光谱位移一致(图10a)。此外,在530ev到535ev之间,特别是在充电超过210mah/g之后,峰值强度增加,这表明在充电时将空穴引入到氧的2p态。
图10d显示了tey模式的氧的k边缘光谱。与tfy模式的氧的k边缘光谱不同,充电过程中约528.5ev处的峰值增长可以忽略不计。相反,在530ev到535ev之间观察到强度增长,如tfy模式的氧的k边缘光谱。这表明,在表面处,ni2++没有被氧化太多,并且主要发生氧气氧化,这与tey模式下镍的l边缘光谱的可以忽略不计的变化相一致。
例2
合成了如下氟取代的阳离子无序锂金属氧化物:
li-ni-ti-mo-o-f型,通式为:li1+xniatibmo1-x-a-bo2-yfy,0.05<x<0.3,0.2<a<0.6,0.2<b<0.6,0<y≤0.3,其中x=0.25,a=0.35,b=0.3,y=0.25(li1.25ni0.35ti0.3mo0.1o1.75f0.25)(“本发明的l125ntmof”)。
本发明的l125ntmof是使用与实施例1中所述相同的前体和程序合成的。
图11a显示了本发明的l125ntmof的x射线衍射(“xrd”)图案。图11b显示了本发明的125ntmof在室温下以20ma/g在1.5和4.5v之间循环时的电压曲线。如图11b所示,本发明的l125ntmof提供184mah/g的第一放电容量和573wh/kg的能量密度。
例3
合成了如下本发明的氟取代的阳离子无序锂金属氧化物:
li-ni-nb-o-f型,通式为:li1+xnianbbo2-yfy,0.05<x<0.3,0.2<a<0.6,0.2<b<0.6,0<y≤0.3,其中x=0.2,a=0.5,b=0.3,y=0.3(li1.2ni0.5nb0.3o1.7f0.3)(“本发明的l120nnof”)。
图12a显示了本发明的l120nnof的x射线衍射(“xrd”)图。图12b显示了当在室温下以20ma/g在1.5和4.5v之间循环时,本发明的l120nnof的电压曲线。如图12b所示,本发明的l120nnof提供325mah/g的第一放电容量和845wh/kg的能量密度。
尽管阐述本发明的广泛范围的数值范围和参数是近似值,但是尽可能精确地给出具体实施例中阐述的数值。然而,任何数值本身都包含一定的误差,这些误差必然是由在各自的测试测量中发现的标准偏差引起的。此外,当在此阐述变化范围的数值范围时,可以考虑使用包括所述值的这些值的任何组合。
本文描述和要求保护的发明不限于本文公开的具体实施方案的范围,因为这些实施方案旨在说明本发明的几个方面。任何等同的实施方案都在本发明的范围内。实际上,除了本文所示和所述的那些之外,本发明的各种修改对于本领域技术人员来说将从前面的描述中变得显而易见,这些修改也旨在落入所附权利要求的范围内。上述文本中引用的所有专利和专利申请均以引用的方式全部明确并入本文。
- 还没有人留言评论。精彩留言会获得点赞!