燃料电池阳极催化剂及其制备方法和质子交换膜燃料电池与流程
本发明属于燃料电池催化剂技术领域,具体涉及一种燃料电池阳极催化剂及其制备方法和质子交换膜燃料电池。
背景技术:
随着科学技术的发展和人类生活水平的提高,人们对石化燃料的消耗与日俱增,在消耗石化燃料的同时也对生态环境造成巨大的破坏。因此,为了解决能源危机,缓解生态环境压力,寻找可再生的替代能源已经刻不容缓。质子交换膜燃料电池具有低温性能好、能量转化率高、结构简单、对环境污染小等诸多优点,被公认为是清洁能源汽车、分散式发电站的首选化学电源,有望在未来大规模开发和应用。目前大多数的质子交换膜燃料电池阳极催化剂的材料是铂等贵金属,其负载量在10wt%以上,会导致成本居高不下,不利于推广应用。另外,阳极催化剂的催化活性低、耐一氧化碳(co)中毒性、稳定性差等也阻碍了其大规模商业化应用,因此有必要寻找一种低成本、高性能的质子交换膜燃料电池阳极催化剂。
技术实现要素:
针对目前质子交换膜燃料电池中铂等贵金属催化剂负载量过多导致质子交换膜燃料电池成本高,并且阳极催化剂活性低、耐一氧化碳中毒性能差、稳定性有待提高等问题,本发明提供一种燃料电池阳极催化剂及其制备方法。
进一步地,本发明还提供包含本发明燃料电池阳极催化剂的质子交换膜燃料电池。
为实现上述发明目的,本发明的技术方案如下:
一种燃料电池阳极催化剂,所述燃料电池阳极催化剂为碳化钼@空心碳球、铂&碳化钼@空心碳球中的至少一种;
其中,碳化钼@空心碳球表示空心碳球表面负载有碳化钼纳米颗粒;
铂&碳化钼@空心碳球表示空心碳球表面同时负载有碳化钼纳米颗粒和铂纳米颗粒。
相应地,一种燃料电池阳极催化剂的制备方法,包括以下步骤:
将氨水溶于乙醇和去离子水的混合溶剂中,得到碱性混合溶剂;
将硅酸乙酯加入所述碱性混合溶剂中,得到混合溶液;
向所述混合溶液中加入盐酸多巴胺,混匀后加入四水合钼酸铵,恒温蒸干,得到前躯体材料;
对所述前躯体材料进行热处理,自然冷却至室温后通入氧气进行钝化处理;钝化处理结束采用氢氟酸进行浸渍处理,得到碳化钼@空心碳球;
采用浸渍法对所述碳化钼@空心碳球进行铂的浸渍负载处理,得到铂&碳化钼@空心碳球。
进一步地,一种质子交换膜燃料电池,包括阳极,所述阳极上含有阳极催化剂,所述阳极催化剂选自如上所述的燃料电池阳极催化剂;或者所述阳极催化剂由如上所述的燃料电池阳极催化剂的制备方法制备得到。
本发明的技术效果为:
相对于现有技术,本发明上述提供的燃料电池阳极催化剂中,碳化钼负载在空心碳球表面形成核壳结构,在催化氢气氧化过程中,加速氢气氧化过程中质子、电子的转移速率从而提高电催化性能。空心碳球结构增加了载体的比表面积,为电催化活性物质提供更多的负载位点,均匀分散在空心碳球上的碳化钼为铂纳米颗粒提供了大量特殊的负载位点,而与碳化钼同时负载在空心碳球表面的铂纳米颗粒尺寸在2~5nm,尺寸均一,分布均匀,在使铂负载量降低的同时,还使得阳极催化剂的比表面积达到760m2/g以上,暴露出更多催化氢气氧化的活性位点,有利于提高阳极催化剂的催化活性,在结合碳化钼之后,可以提高阳极催化剂的耐一氧化碳中毒特性,同时提高阳极催化剂的稳定性。
本发明燃料电池阳极催化剂的制备方法,制备过程简单,原料廉价易得,同时还有效降低阳极催化剂的铂载量,使燃料电池催化剂成本大大降低,适于大规模商业化生产。
本发明的质子交换膜燃料电池与现有的质子交换膜燃料电池相比,由于本发明的阳极催化剂含有碳化钼@空心碳球、铂&碳化钼@空心碳球中的至少一种,其催化活性、耐co中毒性、稳定性等均有大幅度提高,有利于质子交换膜燃料电池的大规模商业化。
附图说明
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
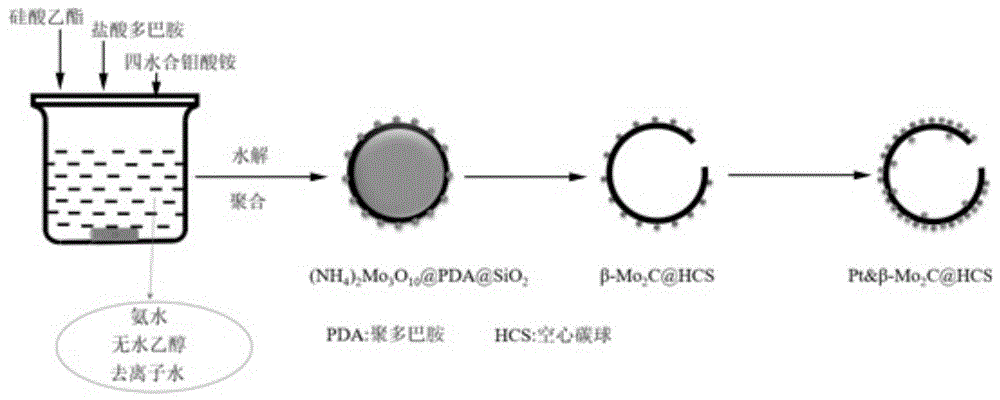
图1为本发明燃料电池阳极催化剂的制备方法工艺流程示意图;
图2为本发明实施例1燃料电池阳极催化剂的制备方法制备的碳化钼@空心碳球的x射线粉末衍射曲线图;
图3为本发明实施例1燃料电池阳极催化剂的制备方法制备的铂&碳化钼@空心碳球的扫描电子显微镜图;
图4为本发明实施例1燃料电池阳极催化剂的制备方法制备的铂&碳化钼@空心碳球的能谱图;
图5为本发明实施例1燃料电池阳极催化剂的制备方法制备的铂&碳化钼@空心碳球的氮气吸附-脱附等温曲线;
图6为本发明实施例2燃料电池阳极催化剂的制备方法制备的铂&碳化钼@空心碳球的扫描透射电子显微镜(stem)图;
图7为本发明实施例1制备的铂&碳化钼@空心碳球质子交换膜燃料电池阳极催化剂的循环伏安曲线;
图8为本发明实施例2制备的铂&碳化钼@空心碳球质子交换膜燃料电池阳极催化剂的循环伏安曲线;
图9为本发明实施例1制备的铂&碳化钼@空心碳球质子交换膜燃料电池阳极催化剂与对比例1的铂碳催化剂的线性扫描伏安曲线;
图10为本发明实施例2制备的铂&碳化钼@空心碳球质子交换膜燃料电池阳极催化剂与对比例1的铂碳的线性扫描伏安曲线。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
本发明中涉及的“逐滴加入”或者“逐滴滴入”,均属于化学技术领域的技术人员熟知的实验操作手段,其滴加的速度、滴加的量均是化学实验的常规操作。
本发明的一方面,提供一种燃料电池阳极催化剂。
所述燃料电池阳极催化剂为碳化钼@空心碳球、铂&碳化钼@空心碳球中的至少一种;
其中,碳化钼@空心碳球表示空心碳球表面负载有碳化钼纳米颗粒;
铂&碳化钼@空心碳球表示空心碳球表面同时负载有碳化钼纳米颗粒和铂纳米颗粒。
下面对本发明的技术方案做进一步的详细解释。
本发明的燃料电池阳极催化剂,可以是只含有碳化钼@空心碳球,也可以只含有铂&碳化钼@空心碳球,或者同时含有碳化钼@空心碳球与铂&碳化钼@空心碳球,减少了对铂的依赖,降低了阳极催化剂中铂的用量,并且具有良好的催化活性。
上述碳化钼@空心碳球中碳化钼纳米颗粒的负载量为10wt%~30wt%,碳化钼具有良好的抗一氧化碳中毒性能,可以有效地实现催化加氢、催化脱氢、电解水的还原等过程,将碳化钼纳米颗粒负载在空心碳球颗粒表面,提高了碳化钼的比表面积,同时还能够使得碳化钼的厚度变薄,在减少碳化钼用量的同时,使得碳化钼的催化活性面积大幅度提高。
上述铂&碳化钼@空心碳球中,铂纳米颗粒的负载量为1.0wt%~5.0wt%,碳化钼纳米颗粒的负载量为10wt%~30wt%,将铂纳米颗粒和碳化钼纳米颗粒同时负载在空心碳球表面,一方面,使得阳极催化剂耐一氧化碳中毒性能得到提高,另一方面,降低了铂的含量,能使得燃料电池阳极催化剂的成本大幅度降低,有利于燃料电池的大规模商业化应用。此外,空心碳球使得铂纳米颗粒和碳化钼纳米颗粒分散更为均匀,暴露出更多的活性位点,碳化钼纳米颗粒和铂纳米颗粒产生协同效应增强催化剂催化活性,获得比商业化铂碳催化剂更好的催化效果。
本发明上述燃料电池阳极催化剂的比表面积达到760m2/g以上,且具有催化活性的碳化钼、铂分散得更加均匀、轻薄,有效催化面积更大,单位面积催化活性更好。
优选地,上述燃料电池阳极催化剂中,空心碳球的粒径为(100~300)nm,厚度为(10~50)nm;碳化钼纳米颗粒的粒径为(1~5)nm;铂纳米颗粒的粒径为(2~5)nm,这样的粒径比例关系,极大的提高了单位面积的催化活性。
本发明第二方面,还提供了上述燃料电池阳极催化剂的制备方法。
请参阅图1,该燃料电池阳极催化剂的制备方法包括以下步骤:
步骤s01.将氨水溶于乙醇和去离子水的混合溶剂中,得到碱性混合溶剂;
步骤s02.将硅酸乙酯加入所述碱性混合溶剂中,得到混合溶液;
步骤s03.向所述混合溶液中加入盐酸多巴胺,混匀后加入四水合钼酸铵,恒温蒸干,得到前躯体材料(该前躯体材料的成分为:(nh4)2mo3o10@pda@sio2);
步骤s04.对所述前躯体材料进行热处理,自然冷却至室温后通入氧气进行钝化处理;钝化处理结束采用氢氟酸进行浸渍处理,得到碳化钼@空心碳球(β-mo2c@hcs);
步骤s05.采用浸渍法对所述碳化钼@空心碳球进行铂的浸渍负载处理,得到铂&碳化钼@空心碳球。
上述步骤s01中涉及的乙醇和去离子水的混合溶剂,是将无水乙醇和去离子水进行混合得到。具体可以将无水乙醇和去离子水按照体积比为(9~11):(31~33)进行混合,混合的过程中,室温即可完成。混合溶剂中乙醇和去离子水的体积比例,有利于避免后续加入的材料发生团聚,并且提高反应过程中产物粒径的均匀性以及分散性。使用的氨水,其质量浓度为(25~28)%(试剂瓶瓶标浓度),氨水与乙醇、去离子水的加料量按体积比为(3~5):(90~110):(310~330)。溶解时,室温即可。
上述所使用的硅酸乙酯的纯度≥98%,密度约为0.93g/ml。将硅酸乙酯加入碱性溶剂中,有利于硅酸乙酯的水解,获得混合溶液。由硅酸乙酯、氨水、乙醇、去离子水四种组分形成的混合溶液中,按照体积比,硅酸乙酯:氨水:乙醇:去离子水=(3~5):(3~5):(90~110):(310~330),当将盐酸多巴胺及四水合钼酸铵加入在该比例范围内得到的混合溶液中时,有利于获得粒径小且均一的前躯体材料。
步骤s03中,将盐酸多巴胺加入上述混合溶液时,可以使得盐酸多巴胺发生氧化沉淀。但是如果盐酸多巴胺的加入速度过快,形成的颗粒不可控且粒径不均匀,不利于形成粒径均一的前躯体材料。为了能够获得粒径均一且细小的前躯体材料颗粒,在盐酸多巴胺加入过程中,有必要控制每(2~3)分钟内盐酸多巴胺的加入量为(500~1000)mg;在每(2~3)分钟的时间长度内均匀加入,避免一次性加入。在加入过程中,为了使得盐酸多巴胺更加均匀地进入混合溶液,可以将盐酸多巴胺制成盐酸多巴胺溶液,如可以制成浓度为(80~100)mg/ml的盐酸多巴胺水溶液,以溶液的方式加入至上述混合溶液中,从而可以保证均匀加入。滴加过程中,还有必要使用机械扰动,如搅拌、超声震荡等等。当用搅拌的方式促进盐酸多巴胺溶液与混合溶液进行混合时,搅拌速度可以是(400~800)rpm,搅拌时长为(24~36)h,室温搅拌即可。
上述盐酸多巴胺溶液的浓度可以是80mg/ml、83mg/ml、85mg/ml、90mg/ml、95mg/ml、98mg/ml、100mg/ml等在80~100mg/ml范围内的任一浓度。
将四水合钼酸铵加入时,有必要采用水浴或者油浴对混合溶液进行加热,加热至(60~80)℃后,开始加入四水合钼酸铵。但是如果四水合钼酸铵的加入速度过快,其在聚多巴胺表面形成的颗粒不可控且粒径不均匀,不利于形成粒径均一的前躯体材料。为了能够获得粒径均一且细小的前躯体材料颗粒,在四水合钼酸铵加入过程中,有必要控制每(2~3)分钟内四水合钼酸铵的加入量为(500~1000)mg;在每(2~3)分钟的时间长度内均匀加入,避免一次性加入。在加入过程中,为了使得四水合钼酸铵进入混合溶液的量更为均匀,可以将四水合钼酸铵制成四水合钼酸铵溶液,如可以制成浓度为(80~100)mg/ml的四水合钼酸铵水溶液,以液体的方式加入至上述混合溶液中,从而可以保证均匀加入。滴加过程中,还有必要使用机械扰动,如搅拌、超声震荡等等。当用搅拌的方式促进四水合钼酸铵溶液均匀分散于盐酸多巴胺氧化产物的表面时,搅拌速度可以是(200~400)rpm,搅拌时长为(6~24)h,在恒温(60~80)℃的条件下搅拌至干燥即可。
上述四水合钼酸铵溶液的浓度可以是80mg/ml、82mg/ml、85mg/ml、88mg/ml、90mg/ml、92mg/ml、95mg/ml、98mg/ml、100mg/ml等在80~100mg/ml范围内的任一浓度。
将盐酸多巴胺、四水合钼酸铵加入混合溶液中时,以混合溶液中硅酸乙酯的质量为参考,各组分的投料质量比为硅酸乙酯:盐酸多巴胺:四水合钼酸铵=(12~25):(5~10):(5~10),盐酸多巴胺及四水合钼酸铵投料量过多,则容易出现团聚,不利于获得粒径小且尺寸均一的前躯体材料。
步骤s04中,热处理时需要在氢气和氩气(h2/ar)氛围中,通过热处理的方式,前躯体材料发生还原获得碳球,并且碳化钼在碳球表面生成。
优选地,热处理的升温速率为(2~10)℃/min,升温至(750~850)℃,保温时间为(2~3)h。在该升温速率的条件下,可以获得在室温下稳定存在、具有六方密堆积结构、尺寸均一、分布均匀的β-mo2c。自然冷却处理的过程中也有必要在氢气和氩气(h2/ar)氛围下进行。
冷却至室温后,通入的氧气可以是纯氧气,也可以是空气,只要携带有氧气的气体均可以通入,其目的是利用氧气对产物进行钝化处理,钝化处理的时长为(2~3)h。
对钝化后的产物进行氢氟酸浸渍时,氢氟酸质量浓度较高(试剂瓶瓶标浓度≥40%),可以先将钝化后的产品溶于去离子水中再滴入氢氟酸溶液,以避免氢氟酸浓度过高而不利于控制浸渍过程。或者按照氢氟酸与去离子水的投料体积比为(3~5):(30~45)进行混合后,将钝化后的产物溶于其中,浸渍时长为6~12h。氢氟酸浸渍时,氢氟酸溶液与钝化产物的投料比例为(11~15):(1~2),氢氟酸投料量过少,无法彻底浸渍刻蚀,无法获得碳化钼@空心碳球,而投料量过多,造成浪费。
在进行步骤s05的铂浸渍负载处理之前,还包括采用去离子水、无水乙醇对碳化钼@空心碳球进行充分清洗、抽滤,并干燥。
步骤s05中,铂的浸渍负载处理使用氯铂酸溶液,浓度为(5~15)mg/ml。由于加入的氯铂酸溶液量较少,不便于进行超声、混合以及浸渍,可以将氯铂酸溶液与去离子水、无水乙醇进行混合后再进行浸渍负载,从而有利于超声分散,有利于铂的均匀负载,同时有利于快速干燥。按照氯铂酸:去离子水:无水乙醇:碳化钼@空心碳球=(1~4):(1000~2000):(1400~2400):(20~40)。尤其控制碳化钼@空心碳球和氯铂酸的投料质量比为(20~40):(1~4),有利于获得粒径均匀的铂纳米颗粒。浸渍过程中超声处理,超声处理的时间为(10~20)min。随后进行干燥处理,干燥后置于h2/ar气氛中按照(3~6)℃/min的升温速率升温至(200~400)℃,恒温(2~3)h,即可获得铂&碳化钼@空心碳球。
当只要获得碳化钼@空心碳球时,至制备步骤s04即可结束。
经过步骤s04获得的碳化钼@空心碳球,比表面积≥760m2/g,碳化钼纳米颗粒的负载量为10wt%~30wt%。所述空心碳球的粒径为(100~300)nm,厚度为(10~50)nm;所述碳化钼纳米颗粒的粒径为(1~5)nm。
经过步骤s05后获得的铂&碳化钼@空心碳球中,铂纳米颗粒的负载量为1.0wt%~5.0wt%,碳化钼纳米颗粒的负载量为10wt%~30wt%;比表面积≥760m2/g,其中空心碳球的粒径为(100~300)nm,厚度为(10~50)nm;所述碳化钼纳米颗粒的粒径为(1~5)nm;所述铂纳米颗粒的粒径为(2~5)nm。
本发明的制备方法工艺简单、原料廉价易得,还能降低铂的载量,可以制备得到粒径均一、负载稳定的阳极催化剂,得到的阳极催化剂具有良好的催化活性和较大的比表面积。此外,本制备方法对设备要求低,适合规模化生产。
本发明获得的燃料电池阳极催化剂可以加入到燃料电池阳极中,从而使得阳极具有良好的催化效果。因此本发明还进一步提供燃料电池阳极以及一种质子交换膜燃料电池。
该燃料电池阳极包括阳极,阳极上负载有阳极催化剂,该阳极催化剂为本发明的燃料电池阳极催化剂。也就是说,本发明的燃料电池阳极的阳极上可以含有碳化钼@空心碳球或者铂&碳化钼@空心碳球,或者同时含有碳化钼@空心碳球和铂&碳化钼@空心碳球。
该质子交换膜燃料电池包括阳极,阳极上含有阳极催化剂,所述阳极催化剂为发明的燃料电池阳极催化剂,具体来说就是本发明质子交换膜燃料电池的燃料电池阳极的阳极上可以含有碳化钼@空心碳球或者铂&碳化钼@空心碳球,或者同时含有碳化钼@空心碳球和铂&碳化钼@空心碳球。
为更有效的说明本发明的技术方案,下面通过多个具体实施例说明本发明的技术方案。
实施例1
一种燃料电池阳极催化剂的制备方法,包括如下步骤:
(a).取500ml的烧杯一个,依次往烧杯加入160ml的去离子水和48ml的无水乙醇,在600rpm的转速下室温搅拌15min,混合均匀,获得混合溶剂。
(b).将2ml质量浓度为25%~28%的氨水逐滴滴入上述混合溶剂中,在600rpm的转速下室温搅拌30min,混合均匀,获得碱性混合溶液。
(c).将2ml纯度≥98%的硅酸乙酯逐滴滴入上述碱性混合溶液,在600rpm的转速下室温搅拌30min,混合均匀,然后再将8ml配制好的浓度为100mg/ml的盐酸多巴胺溶液滴入,控制每(2~3)分钟内盐酸多巴胺进入所述碱性混合溶液的量为(500~800)mg;600rpm的转速下室温搅拌36h,充分反应。
(d).将上述(c)的反应液转移至恒温油浴锅中,加热至80℃,恒温,向其中滴加8ml配制好的浓度为100mg/ml的四水合钼酸铵溶液,控制每(2~3)分钟内四水合钼酸铵进入所述反应液的量为(500~800)mg,恒温的情况下,300rpm的转速搅拌蒸干12h,得到前驱体材料。
(e).将得到的前驱体材料研磨后放入石英舟,然后放入管式炉中,通入h2/ar混合气体,设定升温程序为升温速率为:5℃/min,恒温温度为:800℃,热处理的时间为:2h,随炉冷却至室温,在空气中钝化2h,取出样品。将样品全部倒入100ml的聚四氟乙烯反应釜内衬,先加入80ml去离子水,然后加入8ml质量浓度≥40%的氢氟酸、混合均匀,浸渍6h。将浸渍后的样品抽滤清洗,先用去离子水清洗3次,然后用无水乙醇清洗2次。将清洗后的样品,放入温度恒定为60℃的鼓风干燥箱里,烘干6h,得到碳化钼@空心碳球材料。
(f).取容量为100ml的烧杯,称量100mg步骤(e)得到的碳化钼@空心碳球材料,加入5ml的去离子水,然后加入0.54ml溶度为10mg/ml的氯铂酸溶液,控制每(2~3)分钟内氯铂酸的加入量为(50~100)mg,再加入10ml无水乙醇,将混合液烧杯转移至超声波清洗机,超声10min,放入温度恒定为60℃的鼓风干燥箱里,烘干6h。将干燥后的样品研磨后放入石英舟,然后放入管式炉中,通入h2/ar混合气体,设定升温程序,升温速率为:5℃/min,恒温温度为:300℃,热处理的时间为2h,随炉冷却至室温,得到铂&碳化钼@空心碳球材料。经测试,获得的铂&碳化钼@空心碳球材料的比表面积为761.17m2/g、铂的负载量为1.63wt%、碳化钼的负载量为26.49wt%。
实施例2
一种燃料电池阳极催化剂的制备方法,包括如下步骤:
(a).取容量为500ml的烧杯1个,依次往烧杯加入160ml的去离子水和48ml的无水乙醇,在600rpm的转速下室温搅拌15min,混合均匀,获得混合溶剂。
(b).将2ml质量浓度为25%~28%的氨水逐滴滴入上述混合溶剂中,在600rpm的转速下室温搅拌30min,混合均匀,获得碱性混合溶液。
(c).将2ml纯度≥98%的硅酸乙酯逐滴滴入碱性混合溶液,在600rpm的转速下室温搅拌30min,混合均匀,然后再将8ml配制好的浓度为100mg/ml的盐酸多巴胺溶液,控制每(2~3)分钟内盐酸多巴胺进入所述碱性混合溶液的量为(500~800)mg;600rpm的转速下室温搅拌36h,充分反应。
(d).将上述(c)的反应液转移至恒温油浴锅中,加热至80℃,恒温,向其中滴加8ml配制好的浓度为100mg/ml的四水合钼酸铵溶液,控制每(2~3)分钟内四水合钼酸铵进入所述反应液的量为(500~800)mg,恒温的条件下300rpm的转速搅拌蒸干12h,得到前驱体材料。
(e).将得到的前驱体材料研磨后放入石英舟,然后放入管式炉中,通入h2/ar混合气体,设定升温程序为升温速率为:5℃/min,恒温温度为:800℃,热处理的时间为:2h,随炉冷却至室温,在空气中钝化2h,取出样品。将样品全部倒入100ml的聚四氟乙烯反应釜内衬,先加入80ml去离子水,然后加入8ml质量浓度≥40%的氢氟酸、混合均匀,浸渍6h。将浸渍后的样品抽滤清洗,先用去离子水清洗3次,然后用无水乙醇清洗2次。将清洗后的样品,放入温度恒定为60℃的鼓风干燥箱里,烘干6h,得到碳化钼@空心碳球材料。
(f).取容量为100ml的烧杯,称量100mg步骤(e)得到的碳化钼@空心碳球材料,加入5ml的去离子水,然后加入1.11ml溶度为10mg/ml的氯铂酸溶液,控制每(2~3)分钟内氯铂酸的加入量为(50~100)mg,再加入10ml无水乙醇,将混合液烧杯转移至超声波清洗机,超声10min,放入温度恒定为60℃的鼓风干燥箱里,烘干6h。将干燥后的样品研磨后放入石英舟,然后放入管式炉中,通入h2/ar混合气体,设定升温程序,升温速率为:5℃/min,恒温温度为:300℃,热处理的时间为:2h,随炉冷却至室温,得到铂&碳化钼@空心碳球材料。经测试,获得的铂&碳化钼@空心碳球材料的比表面积为785.71m2/g、铂的负载量为3.86wt%、碳化钼的负载量为18.91wt%。
实施例3
一种燃料电池阳极催化剂的制备方法,包括如下步骤:
(a).取容量为500ml的烧杯1个,依次往烧杯加入150ml的去离子水和50ml的无水乙醇,在600rpm的转速下室温搅拌15min,混合均匀,获得混合溶剂。
(b).将2ml质量浓度为25%~28%的氨水逐滴滴入水醇混合溶剂中,在600rpm的转速下室温搅拌30min,混合均匀,获得碱性混合溶液。
(c).将2ml纯度≥98%的硅酸乙酯逐滴滴入碱性混合溶液,在600rpm的转速下室温搅拌30min,混合均匀,然后再将8ml配制好的浓度为100mg/ml的盐酸多巴胺溶液,控制每(2~3)分钟内盐酸多巴胺进入所述碱性混合溶液的量为(500~800)mg;600rpm的转速下室温搅拌24h,充分反应。
(d).将上述(c)的反应液转移至恒温油浴锅中,加热至80℃,恒温,向其中滴加8ml配制好的浓度为100mg/ml的四水合钼酸铵溶液,控制每(2~3)分钟内四水合钼酸铵进入所述反应液的量为(500~800)mg;恒温条件下,以300rpm的转速搅拌蒸干12h,得到前驱体材料。
(e).将得到的前驱体材料研磨后放入石英舟,然后放入管式炉中,通入h2/ar混合气体,设定升温程序为升温速率为:5℃/min,恒温温度为:800℃,热处理的时间为:3h,随炉冷却至室温,在空气中钝化3h,取出样品。将样品全部倒入100ml的聚四氟乙烯反应釜内衬,先加入85ml去离子水,然后加入8ml质量浓度≥40%的氢氟酸、混合均匀,浸渍12h。将浸渍后的样品抽滤清洗,先用去离子水清洗3次,然后用无水乙醇清洗2次。将清洗后的样品,放入温度恒定为60℃的鼓风干燥箱里,烘干6h,得到碳化钼@空心碳球材料。
(f).取容量为100ml的烧杯,称量100mg步骤(e)得到的碳化钼@空心碳球材料,加8ml的去离子水,然后加入0.54ml溶度为10mg/ml的氯铂酸溶液,控制每(2~3)分钟内氯铂酸的加入量为(50~100)mg,再加入10ml无水乙醇,将混合液烧杯转移至超声波清洗机,超声10min,放入温度恒定为60℃的鼓风干燥箱,烘干6h。将干燥后的样品研磨后放入石英舟,然后放入管式炉中,通入h2/ar混合气体,设定升温程序,升温速率为:5℃/min,恒温温度为:300℃,热处理的时间为3h,随炉冷却至室温,得到铂&碳化钼@空心碳球材料。经测试,获得的铂&碳化钼@空心碳球材料的比表面积为776.22m2/g、铂的负载量为1.84wt%、碳化钼的负载量为27.42wt%。
为了验证上述实施例1~3得到的材料的特性,下面对其进行相关的性能测试。
(一)x射线粉末衍射曲线图
采用bruker,d8advance衍射仪对实施例1的步骤(e)制备得到的碳化钼@空心碳球材料xrd扫描,结果如图2所示。
从图2可知,实施例1得到的碳化钼@空心碳球材料的xrd峰与pdf#77-0720卡片完全吻合,说明制备得到的β-碳化钼物相纯度高,无杂质。
(二)扫描电子显微镜图(sem)
采用tescammira3设备对实施例1步骤(f)制备得到的铂&碳化钼@空心碳球材料进行sem扫描,结果如图3所示。
从图3可看到实施例1得到的材料中空心球表面附着有纳米级的颗粒物。
(三)能谱图(eds)
采用tescammira3设备对实施例1步骤(f)得到的铂&碳化钼@空心碳球材料进行能谱(eds)扫描,结果如图4所示。
从图4可知,铂的负载量为1.63wt%、碳化钼的负载量为26.49wt%,碳含量为71.88wt%。
(四)氮气吸脱附等温曲线
采用日本bel公司belsorp-max型设备对实施例1步骤(f)得到的铂&碳化钼@空心碳球材料进行氮气吸脱附等温曲线测试,结果如图5所示。
从图5可知,实施例1得到的铂&碳化钼@空心碳球材料比表面积为761.17m2/g,而且材料比较符合iv(a)类吸附,有较多的介孔特性,获得的材料为微孔介材料。
(五)stem扫描
采用fei,themisg2设备对实施例2步骤(f)得到的铂&碳化钼@空心碳球材料进行stem及能谱(eds)扫描,结果如图6所示。
从图6可以看到,碳球表面负载有钼、铂颗粒,并且均为纳米级,并且铂的负载量为3.86wt%、碳化钼的负载量为18.91wt%,碳含量为77.23wt%。
(六)循环伏安曲线
将实施例1、2获得的材料分别制成电极,然后采用辰华chi760e型电化学工作站进行循环伏安曲线扫描,同时与对比例1进行对比。
具体地,实施例1、2电极的制作方法如下:
将每个实施例得到的铂&碳化钼@空心碳球3mg,溶于0.4ml异丙醇和0.2ml去离子水溶液中,超声10min,用移液枪量取30μl的5wt%的nafion溶液,继续冰浴超声分散30min。取6μl涂覆于直径为5mm的玻碳电极上,自然晾干,分别制成工作电极,以标准氢电极为参比电极,碳棒为对电极,组成三电极电化学体系,在0.5mh2so4溶液中,分别在通氮气和氢气条件下,进行电化学性能测试。
对比例1的电极为商业化的铂碳电极(jm公司pt/c),该电极中铂的负载量为20wt%。通氮气条件下,电压扫描范围:(0~0.5)v,扫描速率:20mv/s;
通氢气条件下,电压扫描范围:(0~0.5)v,扫描速率:10mv/s。
结果如图7、8、9、10。从图7、8可以看出,制备的材料具有较高的活性比表面积;从图9、10可以看出制备的材料的催化活性明显高于对比例1的商业化铂碳催化剂的催化活性。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包括在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!