用于燃料电池的多元素催化剂结构的制作方法
1.本发明涉及用于燃料电池,例如聚合物电解质膜燃料电池的多元素催化剂结构。
背景技术:
2.燃料电池已经显示出有望成为车辆和其它运输应用的替代动力源。燃料电池通过可再生能源载体如氢气来工作。燃料电池还在没有有毒排放或温室气体的情况下工作。广泛采用和使用这种清洁且可持续技术的当前限制之一是所述燃料电池堆相对昂贵的成本。燃料电池的阳极和阴极二者的催化剂层中都包含催化剂材料(例如铂催化剂材料)。所述催化剂材料是燃料电池堆中最昂贵的组件之一。
技术实现要素:
3.根据一个实施方案,公开了多元素催化剂结构。所述多元素催化剂结构包括由第一金属材料形成的区域、由第二金属材料形成的第一芯部区域和由第三金属材料形成的第二芯部区域。所述第一芯部区域与所述区域具有界面接触。所述第二芯部区域与所述第一芯部区域具有界面接触。所述多元素催化剂结构包含铂(pt)、第一金属m
i
、第二金属m
ii
和第三金属m
iii
。所述第一金属m
i
被配置为增强pt的催化活性。所述第二金属m
ii
被配置为增强所述多元素催化剂结构的稳定性。所述第三金属m
iii
被配置为增强pt、第一金属m
i
、第二金属m
ii
和/或第三金属m
iii
之间的共价结合。
4.根据另一个实施方案,公开了多元素催化剂结构。所述多元素催化剂结构包括由第一金属材料形成的区域、由pt形成的第一芯部区域和由co形成的第二芯部区域。所述第一芯部区域与所述区域具有界面接触。所述第二芯部区域与所述第一芯部区域具有界面接触。
5.在又一个实施方案中,公开了燃料电池。所述燃料电池包括聚合物电解质膜(pem)。所述燃料电池还包括分别包含第一和第二载体以及第一和第二催化剂材料的第一和第二电极催化剂层。pem位于所述第一和第二电极催化剂层之间。所述第一和/或第二催化剂材料包含多个多元素催化剂颗粒。所述多个多元素催化剂颗粒中的每一个包括由第一金属材料形成的区域、由第二金属材料形成的第一芯部区域和由第三金属材料形成的第二芯部区域。所述第一芯部区域与该区域具有界面接触。所述第二芯部区域与所述第一芯部区域具有界面接触。所述多元素催化剂结构包含铂(pt)、第一金属m
i
、第二金属m
ii
和第三金属m
iii
。所述第一金属m
i
被配置为增强pt的催化活性。所述第二金属m
ii
被配置为增强所述多元素催化剂结构的稳定性。所述第三金属m
iii
被配置为增强pt、第一金属m
i
、第二金属m
ii
和/或第三金属m
iii
之间的共价结合。
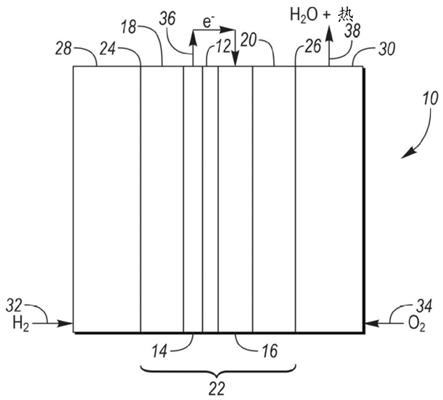
6.附图简述图1是燃料电池的示意性侧视图。
7.图2是计算平台的示意图,其可用于实施一个或多个实施方案的dft算法、计算和/或方法学。
8.图3a至3f描绘了用于燃料电池的不同催化剂层结构的示意图。
9.图4a至图4f描绘了用于燃料电池的co掺杂的pt催化剂模型的示意图。
10.图5a至5j描绘了使用dft计算的催化剂层模型的示意图。
11.图6a描绘了pourbaix图,其示出了金属(锡)
‑
聚合物电解质(pem)体系的热力学平衡态的图形表示。
12.图6b描绘了显示包埋在催化剂层内各个位置处的pt
‑
m催化剂中的十种不同金属(m =钴(co)、镍(ni)、铼(re)、钨(w)、钼(mo)、锗(ge)、锡(sn)、铌(nb)、钽(ta)和钛(ti))的计算dft结果的曲线图。
13.图7描绘了显示若干元素的电负性的周期表。
14.图8a至图8i描绘了被配置为用于根据一个或多个实施方案的燃料电池的电极催化剂层中的多元素催化剂结构的示意性侧视图。
具体实施方式
15.下文描述本公开的实施方案。然而,应当理解,所公开的实施方案仅是实例,并且其它实施方案可以采取各种和替代的形式。附图不必是按比例的;一些特征可能被夸大或最小化,以示出特定组件的细节。因此,本文公开的具体结构和功能细节不应解释为限制性的,而仅仅是作为教导本领域技术人员以各种方式使用实施方案的代表性基础。如本领域普通技术人员将理解的,参考任何一个附图示出和描述的各种特征可以与一个或多个其它附图中示出的特征组合,以产生未明确示出或描述的实施方案。所示特征的组合提供了用于典型应用的代表性实施方案。然而,与本公开的教导一致的特征的各种组合和修改对于特定应用或实施而言可能是所希望的。
16.除了实施例中或另有明确说明外,本说明书中所有表示材料量或反应和/或使用条件的数值应理解为在描述本发明的最宽范围时由词语“约”修饰。在所声明的数字限值内的实践通常是优选的。并且,除非有相反的明确说明:百分比,“份”和比率值均以重量计;术语“聚合物”包括“低聚物”、“共聚物”、“三元共聚物”等;结合本发明作为对给定目的而言合适或优选的材料组或类的描述意味着该组或类的任何两个或更多个成员的混合物同样合适或优选;对于任何聚合物提供的分子量是指数均分子量;用化学术语对成分的描述是指添加到描述中指定的任何组合时的成分,并不一定排除一旦混合之后混合物的成分之间的化学相互作用;首字母缩写词或其它缩写词的第一定义适用于本文中相同缩写词的所有后续使用,并且加以必要修改适用于最初定义的缩写词的常规语法变体;并且,除非有相反的明确说明,否则性能的测量是通过与之前或之后针对相同性能的相同技术来测定的。
17.本发明不限于下面描述的特定实施方案和方法,因为特定的组件和/或条件当然可以变化。另外,本文使用的术语仅以描述本发明的实施方案为目的,并不旨在以任何方式进行限制。
18.本说明书和所附权利要求书中使用的单数形式“一个”、“一种”和“所述”包括多数形式的指示对象,除非上下文中另外有明确说明。例如,以单数形式提及组件旨在包括多个组件。
19.术语“基本上”在本文中可用于描述所公开或要求保护的实施方案。术语“基本上”可以修饰在本公开中所公开或要求保护的值或相对特性。在这种情况下,“基本上”可表示
其修饰的值或相对特性在所述值或相对特性的
±
0%、0.1%、0.5%、1%、2%、3%、4%、5%或10%之内。
20.由于二氧化碳排放量增加以及运输领域中对不可再生化石燃料作为能源载体的当前相对高的依赖性,对开发和商业化使用清洁且可持续能源的运输技术的需求日益增长。一种有前途的技术是燃料电池(包括聚合物电解质膜燃料电池(pemfc))。燃料电池使用来自空气的氧气和压缩氢气作为燃料源,但是仅排放水和热。燃料电池的广泛采用将减少二氧化碳排放。但是,广泛采用需要进一步研究降低燃料电池中使用的催化剂(即铂催化剂)的成本。
21.典型的单燃料电池由聚合物电解质膜(pem)、阳极层、阴极层和气体扩散层构成。这些组件形成膜电极组装件(mea),该膜电极组件被两个流场板围绕。在所有mea组件中,由于必须在阳极和阴极二者处使用铂(pt)且缺乏通过规模效应经济来降低成本的机会,因此在阳极和阴极层二者中发现的催化剂通常是最昂贵的成分。纯pt、pt
‑
co和pt
‑
ni纳米颗粒是工业中pemfc最常用的催化剂材料。
22.铂催化剂经受pt
2+
离子从催化剂层溶解和迁移到燃料电池中的其它组件,如聚合物电解质膜(pem)。在阳极处,pt催化氢气氧化反应(hor,h2ꢀ→ꢀ
2h+ + 2e
‑
);且在阴极处,pt催化氧气还原反应(orr,
½
o
2 + 2h+ + 2e
‑ꢀ→ꢀ
h2o)。阴极处所需的pt负载量显著高于阳极处,因为orr的动力学明显比hor的动力学慢。此外,即使在阳极处使用低至0.025 mgpt/cm2的负载量时,hor仍可以小于20 mv的动力学损失进行。或者,即使在使用0.1
‑
0.4 mgpt/cm2的显著较高的铂负载量时,在与fcv工作相关的电流下,orr的动力学损失也大约为~400 mv。在燃料电池工作过程中,在阳极和阴极二者处,pt都可能进一步降解(例如,溶解、迁移和重新沉积,这会导致电化学活性表面积的损失),这将随后增加整个pemfc堆体系在其整个寿命中经历的动力学过电势。
23.典型地,由于许多不同的可能因素(例如,溢流(flooding)、催化剂降解、由于酸性环境引起的腐蚀、材料失效、过电势积聚等),任何pemfc堆可能随着时间而降解。使pemfc在大于0.8v的较高电压下工作可能对于堆体系产生更多的功率,但是,这可能使得所述催化剂(和其它组件)降解得更快。
24.需要的是在保持铂催化剂的有益活性的同时减少溶解并减慢迁移的解决方案。本公开的各方面涉及作为催化剂的多元素纳米颗粒,以通过产生新型非均相结构化的催化剂来减少降解并增加稳定性来减少活性催化剂的量。非均相结构化的催化剂的使用保持了电化学表面活性面积(esca)和/或减轻了pem降解,从而在给定的催化剂材料负载量下延长了燃料电池堆的寿命。
25.图1描绘了燃料电池10的示意性侧视图。各个燃料电池10可以堆叠以形成燃料电池堆。燃料电池10包括聚合物电解质膜(pem)12、阳极层14、阴极层16以及第一和第二气体扩散层(gdl)18和20。pem 12位于阳极层14和阴极层16之间。阳极层14位于第一gdl 18和pem 12之间,且阴极层16位于第二gdl 20和pem 12之间。pem12、阳极14、阴极16以及第一和第二gdl 18和20构成膜电极组装件(mea)22。mea 22的第一侧24和第二侧26分别由流场28和30界定。流场28向mea 22供应h2,如箭头32所示。流场30向mea 22供应o2,如箭头34所示。在阳极层14和阴极层16中使用催化剂材料如铂。所述催化剂材料通常是mea 22最昂贵的成分。所述催化剂材料负载在催化剂载体上。
26.在一个实施方案中,使用第一性原理密度泛函理论(dft)算法、计算和/或方法学以确定发生在催化剂表面的orr的热力学反应路径。所述dft算法可用于模拟pt催化剂上吸附的氧原子及其还原为oh和与另一h
+
(或h3o
+
)反应,这可生成水分子(h2o)。该模拟解释了每种被吸附物(
‑
h、
‑
oh、
‑
o和h2o)的化学结合和/或物理结合的强度以及它如何影响催化活性和稳定性。
27.一个或多个实施方案的dft算法、计算和/或方法学使用计算机平台,如图2所示的计算平台50来实施。计算平台50可以包括处理器52、存储器54和非易失性存储器56。所述处理器52可包括一个或多个选自高性能计算(hpc)系统的设备,其包括高性能内核、微处理器、微控制器、数字信号处理器、微型计算机、中央处理单元、现场可编程门阵列、可编程逻辑设备、状态机、逻辑电路、模拟电路、数字电路或基于驻留在存储器54中的计算机可执行指令来操纵信号(模拟或数字)的任何其它设备。所述存储器54可包括单个存储器设备或多个存储器设备,其包括但不限于随机存取存储器(ram)、易失性存储器、非易失性存储器、静态随机存取存储器(sram)、动态随机存取存储器(dram)、闪存、高速缓冲存储器或能够存储信息的任何其它设备。所述非易失性存储器56可包括一个或多个持久数据存储设备,如硬盘驱动器、光盘驱动器、磁带驱动器、非易失性固态设备、云存储或能够持久存储信息的任何其它设备。
28.处理器52可被配置为读入存储器54并执行驻留在非易失性存储器56的dft软件模块58中且体现一个或多个实施方案的dft平板模型算法、计算和/或方法学的计算机可执行指令。软件模块58可包括操作系统和应用程序。可以从使用各种编程语言和/或技术创建的计算机程序中编译或解释软件模块58,所述编程语言和/或技术包括但不限于单独或组合的java、c、c++、c#、objective c、fortran、pascal、java script、python、perl和pl/sql。
29.由处理器52执行后,dft软件模块58的计算机可执行指令可以使计算平台50实施本文公开的dft算法和/或方法学中的一种或多种。非易失性存储器56还可包括支持本文描述的一个或多个实施方案的功能、特征、计算和方法的dft数据60。
30.体现本文描述的算法和/或方法学的程序代码能够作为各种不同形式的程序产品单独或集中分发。可以使用其上具有用于使处理器执行一个或多个实施方案的各方面的计算机可读程序指令的计算机可读存储介质来分发程序代码。本质上非暂时性的计算机可读存储介质可包括以用于存储信息,如计算机可读指令、数据结构、程序模块或其它数据的任何方法或技术实施的易失性和非易失性以及可移动和不可移动的有形介质。计算机可读存储介质可进一步包括ram、rom、可擦可编程只读存储器(eprom)、电可擦可编程只读存储器(eeprom)、闪存或其它固态存储技术、便携式光盘只读存储器(cd)
‑
rom)或其它光学存储器、盒式磁带、磁带、磁盘存储器或其它磁性存储设备,或可用于存储所需信息并可由计算机读取的任何其它介质。可以将计算机可读程序指令从计算机可读存储介质下载到计算机、另一类型的可编程数据处理装置或另一设备,或者经由网络下载到外部计算机或外部存储设备。
31.存储在计算机可读介质中的计算机可读程序指令可用于指导计算机、其它类型的可编程数据处理装置或其它设备以特定方式发挥作用,以使得存储在计算机可读介质中的指令产生包括实施流程图或图表中指定的功能、动作和/或操作的指令的制品。在某些替代实施方案中,流程图和图表中指定的功能、动作和/或操作可以与一个或多个实施方案一致
地被重新排序、串行处理和/或同时处理。另外,任何流程图和/或图表可包括相比于与一个或多个实施方案一致地示出的那些而言更多或更少的节点或方框。
32.如由dft软件模块58所生成,图3a至3f描绘了不同催化剂层的示意图以及pt催化剂上的co掺杂和取代对orr反应(即燃料电池的电势)的影响。如dft计算所示,不同的化学掺杂和/或取代可显著影响催化剂活性。dft软件模块58计算了在纯pt和pt
‑
co催化剂材料处发生的orr反应的结果,其中co原子位于pt(111)催化剂平板内的各个位置。图3c描绘了位于pt(111)催化剂平板102的位点100处的co原子。图3d描绘了位于pt(111)催化剂平板102的位点104处的co原子。图3e描绘了位于pt(111)催化剂平板102的位点106处的co原子。图3f描绘了位于pt(111)催化剂平板102的位点108处的co原子。如图3a所示,所述dft计算证明了以下反应在纯pt中在相对于v
rhe
而言0.74 v下发生。
33.催化剂层110描绘了反应(1)的反应物侧,包括铂原子112、氢氧根离子114和氢离子116。催化剂层118描绘了反应(1)的产物侧,包括铂原子112和水分子120。如图3b所示,所述dft计算证明了以下反应在纯pt中在相对于v
rhe
而言0.95 v发生。
34.根据dft计算,当在pt(111)催化剂平板102的近表面中的位点100或位点104处取代co时,反应(1)和(2)的电势增加。根据dft计算,当在位于趋向本体的位点106和108处co取代时,co带来的影响较小。例如,即使pt催化剂中的co浓度不同,位于位点106处的co也显示出与位于位点100处的co相似的电势。在co位于位点108处的情况下,与纯pt相比,该影响可忽略不计。由于位于位点108处的co导致与位于位点106处的co的相同电势,因此纯pt催化剂可用pt
‑
co合金代替,在该pt
‑
co合金中三个pt层位于co金属的顶部(例如,pt壳和co芯)。在这样的实施方案中,包含co的催化剂材料可以节省pt负载量和成本,同时提供相比于纯pt而言相同的性能。下面所示的表1显示了每一类型的pt(111)催化剂(有和没有co)的反应电势:催化剂反应(1)反应(2)标准电势纯pt(111)0.74v0.95v与实验吻合良好co掺杂的近表面(即100)0.84v0.98v由反应(1)增加0.1vco取代(即104)1.07v1.29v由反应(1)和(2)增加
‑
0.3vco取代(即106)0.84v0.97v与(1)类似co取代(即108)0.72v0.93v与纯pt类似表1。
35.取决于刻面(facet)分布、一种或多种合金元素的浓度以及pt催化剂内的合金分布(例如,表面、近表面或趋向本体),催化活性以及稳定性可能会受到显著影响。所述表面层可以是最外面的顶表面层。所述近表面层可以是表面层直接下方的一个层。本体层可以是在表面层和近表面层下方的层。在其它实施方案中,所述表面和近表面层可以是前三层,因为所述表面可能不是原始的。
36.如由dft软件模块58所生成,图4a至4f分别描绘了co掺杂的pt催化剂模型150、152、154、154、156、158和160。催化剂模型150在表面位点162处包含掺杂的co。催化剂模型
152在近表面位点164处包含掺杂的co。催化剂模型154在第三层位点166处包含掺杂的co。催化剂模型156在第四层位点168处包含掺杂的co。催化剂模型158在第五层位点170处包含掺杂的co。催化剂模型160在本体区域172处包含掺杂的co。在另一个实施方案中,所述co掺杂可以用ni掺杂代替。下表2使用计算的dft值报道了co和ni的掺杂能(δe
掺杂
)[ev/位点]。δe
掺杂
[ev/位点]ni掺杂co掺杂表面0.2260.580近表面
‑
0.306
‑
0.001第三层
‑
0.2570.037第四层
‑
0.2670.043第五层
‑
0.2390.054本体
‑
0.1480.138表2。
[0037]
从dft计算的数据观察到,与co掺杂的pt体系相比,对于ni的计算掺杂能(δe
掺杂
)通常更“负”。ni和pt的晶体结构相似,即二者都是面心立方(fcc),而co具有的基态是六方密堆积(hcp)结构。当晶体结构、尺寸和/或电子价相似时,可能更容易诱发混合。所述dft计算的数据还证实了由于ni和pt的强混合,可能更难以在pt催化剂的近表面区域处偏析(segregate) ni原子。当计算的δe
掺杂
小于零时,有利地诱发混合。从dft计算结果表明了,co在pt(111)的近表面处或多或少偏析,而ni可以混合在整个pt中(除了pt的表面处之外)。如表2所示,在表面处ni和co掺杂的相对高的正值证实了趋向pt表面的ni和co偏析是最不利的。表2还显示了ni和co占据近表面是最有利的。在另外层中的掺杂的趋势朝着本体值的方向下降。
[0038]
图5a至图5j使用dft计算分别描绘了催化剂层200、202、204、206、208、210、212、214、216和218。催化剂层200是在表面处具有结合氧原子220的纯pt(111)材料。催化剂层202包含ni掺杂222和表面处的结合氧224。催化剂层204包含近表面区域处的ni原子226和表面处的结合氧228。催化剂层206包含co掺杂230和表面处的结合氧232。催化剂层208包含近表面区域处的co原子234和表面处的结合氧236。催化剂层210是在近表面区域处具有结合氧原子238的纯pt(111)材料。催化剂层212包含ni掺杂240和近表面区域处的结合氧242。催化剂层214包含近表面区域处的ni原子244和近表面区域处的结合氧246。催化剂216包含co掺杂248和近表面区域处的结合氧248。催化剂218包含近表面区域处的co原子252和近表面区域252处的结合氧254。表3显示了所描绘的纯pt、pt
‑
ni和pt
‑
co体系的计算的dft氧结合能(δe
结合,o
)。δe
掺杂
[ev/o]表面近表面pt(111)
‑
1.664+1.374ni掺杂
‑
1.189+0.555ni近表面
‑
1.100
‑
0.407co掺杂
‑
1.146+0.285co近表面
‑
1.055
‑
0.992表3。
[0039]
取决于催化剂的组成,氧结合能可能受到显著影响,从而导致不同的催化活性和
降解。纯pt(111)表面上的δe
结合,o
为
‑
1.664 ev,而ni和co掺杂/取代的pt(111)的δe
结合,o
在
‑
1.0至
‑
1.2 ev之间变化。氧结合能的增加表明,一旦氧原子被吸附在催化剂表面处,它们也可能更容易解吸,从而导致催化活性增加(更容易形成h2o)。纯pt(111)近表面上的δe
结合,o
为+1.374 ev,这表明氧气在近表面处渗透和形成花费大量能量。然而,一旦在pt催化剂处发生co和ni掺杂和取代,δe
结合,o
就显著降低。这表明,在更容易触发pt和co/ni溶解步骤的情况下,尤其是在pemfc的高工作电势下,基底氧化物形成可能变得更有利。尽管pt中的ni和co掺杂和取代可以增强催化活性,但与纯pt催化剂相比,pt
‑
m催化剂的稳定性并不有利。在一个或多个实施方案中,催化活性(即pemfc性能)、催化稳定性(即pemfc寿命)和催化剂成本同时被优化。
[0040]
图6a描绘了pourbaix图260,其显示了锡(sn)的热力学平衡态作为不同酸度和电压的函数的图形。pourbaix图260绘制了以e(v)为单位的锡(sn)的电极电势262相对于电解质的ph 264的图。pourbaix图260显示了,当ph从1变到4且电压从0波动到1v时(确定与pemfc工作有关的工作状况),sn将钝化为sno2。除co、ni和re之外,其它元素(w、mo、ge、sn、nb、ta和ti)可在这一区域处形成氧化物:例如,wo3‑
x
、moo3‑
x
、geo2‑
x
、sno2‑
x
、nb2o5‑
y
、ta2o5‑
y
和tio2‑
x (其中0 ≤ x ≤ 2且0 ≤ y ≤ 5)。在这一pemfc工作状况中,co、ni和re将可能变成co
2+
、ni
2+
和reo4‑
。
[0041]
图6b描绘了显示包埋在催化剂层内各个位置处的pt
‑
m催化剂中的十种不同金属(m=co、ni、铼(re)、钨(w)、钼(mo)、锗(ge)、锡(sn)、铌(nb)、钽(ta)和钛(ti))的计算dft结果的曲线图270。曲线图270通过将作为y轴272的混合能[ev/位点]相对于作为x轴274的距催化剂层表面的距离绘制而显示了pt
‑
m的偏析趋势。每种金属的最低混合能是每种金属曲线上的最低点。通常,dft混合能的正值表明相生成,而负值表明可诱发混合。混合能的大小决定了混合的强或弱。例如,ti和pt的混合比ni和pt的混合更强。这里,如果直接暴露于pefmc环境,co、ni和re可能离子化为co
2+
、ni
2+
、reo4‑
。在pemfc工作状况过程中,其它元素可能钝化,即形成稳定的氧化物mo
x
。
[0042]
如通过本文先前报道的dft计算所示,co和ni在近表面区域中具有最强的优选。co的混合能通常为正(由于其晶体结构不同),而ni优选与pt混合(即混合能略低于零),因为其晶体结构相似。re与co和ni也具有相似大小的混合能,然而,它最优选以本体规模混合。当与ni、co和re相比时,图6b中列出的其它元素的混合相对非常强(即,非常负的混合能)。w和mo具有相似的趋势,其中最优选与pt本体混合。ge和sn享有相同的趋势,其中表面混合是最优选的(而总体混合能是相当负的)。nb和ta在本体处强烈混合,而ti优选留在近表面处。
[0043]
如图6b证实,pt
‑
m中的不同元素m可导致催化剂材料内的不同偏析,所有这些均导致不同的催化活性和稳定性。例如,co和ni都可以提高催化活性;然而,pt
‑
co和pt
‑
ni由于更有利的近表面氧化物形成以及其在pemfc工作过程中一旦在表面处暴露而离子化为co
2+
和ni
2+
的趋势而会导致更快的降解。图6b显示了在pt
‑
sn上的dft计算,其中sn倾向于趋向表面偏析。尽管pt
‑
sn处的氧结合能增加了(不如pt
‑
co或pt
‑
sn那样大),但在pemfc工作过程中,这增加了催化活性,以更易于形成h2o。同时,当与co
2+
或ni
2+
体系相比时,sn仅钝化为sno2,但不易溶解。
[0044]
鉴于图3至6所示的dft计算,选择材料和相互关系可影响pemfc性能、降解和寿命。在一个或多个实施方案中,公开了具有除铂之外的三种或更多种不同金属的多元素催化剂
结构(pt
‑
m
i
‑
m
ii
‑
m
iii
)。所述多元素催化剂结构被配置为用于pemfc的催化剂层中。每个多元素催化剂颗粒的尺寸可以为任何以下值或者在以下值中任两个的范围内:2、3、4、5、6、7、8、9、10、20、30、40、50、100、150、200和250 nm。
[0045]
在某些实施方案中,m
i
是增强催化活性的金属,另外称为催化活性增强剂。m
i
金属包括3d、4d和5d过渡金属,其包括但不限于钒(v)、铬(cr)、锰(mn)、铁(fe)、co、ni、铜(cu)(3d过渡金属);y和zr(4d过渡金属);以及hf和re(5d过渡金属)。m
i
金属被配置为影响pt的电子结构以增加催化活性。当m
i
金属暴露于pemfc环境(ph为1至4;相对于v
rhe
而言的电压为0至1 v)时,所述m
i
金属可被离子化并溶解到溶液中(例如ni
2+
、mn
2+
、fe
3+
、co
2+
等)。
[0046]
在某些实施方案中,m
ii
是有助于多元素催化剂的稳定性的金属,另外也称为稳定性增强剂。m
ii
金属的非限制性实例包括贵金属,如钌(ru)、铑(rh)、钯(pd)、银(ag)、锇(os)、铱(ir)和金(au),以及钝化金属,如w、mo、ge、sn、nb、ta和ti。钝化金属在氧化条件(例如高压和/或高ph)下可变成钝化氧化物,但是在pemfc工作条件下不溶于酸性环境。取决于催化剂的局部环境,w、mo、ge、sn、nb、ta和ti可氧化为wo3‑
x
、moo3‑
x
、geo2‑
x
、sno2‑
x
、nb2o5‑
y
、ta2o5‑
y
和tio2‑
x
(其中0 ≤ x ≤ 2且0 ≤ y ≤ 5)。
[0047]
在某些实施方案中,m
iii
是被配置为在pt、mi,mii和载体材料(例如碳)之间诱发相对强的共价结合的金属。例如,可以通过比较电负性来选择m
iii
金属。图7描绘了显示若干元素的电负性的周期表300。
[0048]
金属键通常具有非常小的电负性差值(如果有的话)。如果电负性差值超过1.7,则极有可能形成离子键。共价键通常在低于1.7下形成。极性共价键通常在0.3至1.7下形成。例如,如果m
i
等于ni且m
ii
等于ge(二者的电负性均为1.8),则选择电负性(χ)为0.1 <χ<1.5或2.1 <χ<3.5的元素。同时,考虑到与pt(χ= 2.2)和c(χ= 2.5)的电负性差值:对于pt为0.5 < χ < 1.9或2.5 < χ < 3.9,且对于c为0.8 < χ < 2.2或2.8 < χ < 4.2。在该实施方案中,电负性为0.8 <χ<1.5或2.8 <χ<3.5的元素可以满足与pt、ni、ge和c形成共价结合的准则。虽然并非很多金属满足该条件的上限(2.8 <χ<3.5),但是一些金属如mg(1.2)、sc(1.3)、y(1.2)、zr(1.4)、hf(1.3)和许多其它金属都可以满足这一准则的下限。选择用于m
iii
的金属可具有对于成为m
i
(增强活性)或m
ii
(增强稳定性)的双重作用。另外,对于除碳之外的催化剂载体如tio2和sno2,基于电负性修改了选择m
iii
的程序。
[0049]
图8a至图8i描绘了被配置为用于根据一个或多个实施方案的燃料电池的电极催化剂层中的多元素催化剂结构350、352、354、356、358、360、362、364和366的示意性侧视图。如图8a至8i所示,多元素催化剂结构350、352、354、356、358、360、362、364和366通常为三角形的。在一个或多个实施方案中,多元素催化剂结构的形状可根据合成条件和元素之间的界面能而变化。形状的非限制性实例通常包括卵形、八面体、三角形、矩形、立方体、六边形及其组合。所述形状可沿着该形状的三个维度中的两个展平。所述多元素催化剂颗粒可包含至少1原子%的m
i
、m
ii
和m
iii
金属。m
i
、m
ii
和m
iii
金属的原子%累计可为小于50原子%。
[0050]
在一个或多个实施方案中,使pt和/或pt
‑
m
ii
的表面积暴露最大化是所希望的,以增加电化学活性表面积(ecsa)。在一个或多个实施方案中,由于m
ii
金属不导致严重的溶解,因此提供了pt基质内的小浓度m
ii
金属的暴露。所述小浓度可以为任何以下值或者在以下值中任两个的范围内:1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16和16.7原子%。尽管m
i
金属可增强催化活性,但m
i
金属可溶于酸(例如ni
2+
、co
2+
)。在其它实施方案中,所述m
i
和m
ii
金属
可在催化剂的芯部区域处一起形成基质。m
iii
金属可与pt形成对催化剂载体材料(例如碳、tio、sno2)具有更强吸引力的分明的界面。在某些实施方案中,增加与其它元素pt、m
i
和m
ii
的界面接触对于更强地形成共价网络结合而言是所希望的。所述多元素催化剂材料在催化剂载体上的负载量可以为任何以下值或者在以下值中任两个的范围内:0.01、0.025、0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9和1.0 mg催化剂/cm2。
[0051]
图8a描绘了负载在催化剂载体层368上的多元素催化剂结构350的示意性侧视图。多元素催化剂结构350包括区域386、第一芯部区域388和第二芯部区域390。区域386的形状通常为球形。第一芯部区域388的形状通常为卵形。第二芯部区域390的形状通常为卵形。区域386的一部分与催化剂载体层368直接接触。区域386被配置为负载第一和第二芯部区域388和390。区域386的一部分与第一芯部区域388和第二芯部区域390界面接触。第一芯部区域388和第二芯部区域390的部分与彼此界面接触。由m
iii
金属形成的区域386与催化剂载体层368和pt以及m
i
和m
ii
金属接触。区域386可以是纯m
iii
金属。在其它实施方案中,区域386由任何以下值或在以下值中任两个的范围内的m
iii
金属制成:90、91、92、93、94、95、96、97、98、99和99.5原子%。第一芯部区域388由pt形成。第一芯部区域388可以是纯pt。在其它实施方案中,第一芯部区域388由任何以下值或在以下值中任两个的范围内的pt制成:90、91、92、93、94、95、96、97、98、99和99.5原子%。第二芯部区域390由m
i
和m
ii
金属的基质形成。在所述第二芯部区域390中m
i
金属与m
ii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。
[0052]
图8b描绘了负载在催化剂载体层370上的多元素催化剂结构352的示意性侧视图。多元素催化剂结构352包括区域392、第一芯部区域394和第二芯部区域396。如图8b所示,区域392由两个不连续区域形成。每个不连续区域的形状通常为球形。第一芯部区域394的形状通常为卵形。第二芯部区域396的形状通常为卵形。区域392的一部分与催化剂载体层370直接接触。区域392被配置为负载第一和第二芯部区域394和396。区域392的一部分与第一芯部区域394和第二芯部区域396界面接触。第一芯部区域394和第二芯部区域396的部分与彼此界面接触。区域392由m
iii
金属形成。区域392可以是纯m
iii
金属。在其它实施方案中,区域392由任何以下值或在以下值中任两个的范围内的m
iii
金属制成:90、91、92、93、94、95、96、97、98、99和99.5原子%。第一芯部区域394的一部分可以与催化剂载体层370直接接触。第一芯部区域394由pt形成。第一芯部区域可以是纯pt。在其它实施方案中,第一芯部区域394由任何以下值或在以下值中任两个的范围内的pt制成:90、91、92、93、94、95、96、97、98、99和99.5原子%。第二芯部区域396的一部分可以与催化剂载体层370直接接触。第二芯部区域396由m
i
和m
ii
金属的基质形成。在所述第二芯部区域396中m
i
金属与m
ii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。
[0053]
图8c描绘了负载在催化剂载体层372上的多元素催化剂结构354的示意性侧视图。多元素催化剂结构354包括区域398、第一芯部区域400和第二芯部区域402。区域398的形状通常为球形。第一芯部区域400的形状通常为卵形。第二芯部区域402的形状通常为卵形。区域398的一部分与催化剂载体层372直接接触。区域398被配置为负载第一和第二芯部区域400和402。区域398的一部分与第一芯部区域400和第二芯部区域402界面接触。第一芯部区域400和第二芯部区域402的部分与彼此界面接触。区域398由m
iii
金属形成。区域398可以是纯m
iii
金属。在其它实施方案中,区域398由任何以下值或在以下值中任两个的范围内的m
iii
金属制成:90、91、92、93、94、95、96、97、98、99和99.5原子%。第一芯部区域400由pt和m
ii
金属的基质形成。在所述第一芯部区域400中pt与m
ii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。第二芯部区域402由m
i
金属形成。第二芯部区域402可以是纯m
i
金属。在其它实施方案中,第一芯部区域402由任何以下值或在以下值中任两个的范围内的m
i
金属制成:90、91、92、93、94、95、96、97、98、99和99.5原子%。
[0054]
图8d描绘了负载在催化剂载体层374上的多元素催化剂结构356的示意性侧视图。多元素催化剂结构356包括区域404、第一芯部区域406和第二芯部区域408。如图8d所示,区域404由两个不连续区域形成。每个不连续区域的形状通常为球形。第一芯部区域406的形状通常为卵形。第二芯部区域408的形状通常为卵形。区域404的一部分与催化剂载体层374直接接触。区域404被配置为负载第一和第二芯部区域406和408。区域404的一部分与第一芯部区域406和第二芯部区域408界面接触。第一芯部区域406和第二芯部区域408的部分与彼此界面接触。区域404由m
iii
金属形成。区域404可以是纯m
iii
金属。在其它实施方案中,区域404由任何以下值或在以下值中任两个的范围内的m
iii
金属制成:90、91、92、93、94、95、96、97、98、99和99.5原子%。第一芯部区域406的一部分可以与催化剂载体层374直接接触。第一芯部区域406由pt和m
ii
金属的基质形成。在所述第一芯部区域406中pt与m
ii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。第二芯部区域408的一部分可以与催化剂载体层374直接接触。第二芯部区域408由m
i
金属形成。第二芯部区域408可以是纯m
i
金属。在其它实施方案中,第一芯部区域408由任何以下值或在以下值中任两个的范围内的m
i
金属制成:90、91、92、93、94、95、96、97、98、99和99.5原子%。
[0055]
图8e描绘了负载在催化剂载体层376上的多元素催化剂结构358的示意性侧视图。多元素催化剂结构358包括区域410、第一芯部区域412和第二芯部区域414。区域410的形状通常为卵形。第一芯部区域412的形状通常为卵形。第二芯部区域414的形状通常为卵形。区域410的一部分与催化剂载体层376直接接触。区域410被配置为负载第一和第二芯部区域412和414。区域410的一部分与第一芯部区域412和第二芯部区域414界面接触。第一芯部区域412和第二芯部区域414的部分与彼此界面接触。区域410由m
ii
和m
iii
金属的基质形成。在所述区域410中m
ii
金属与m
iii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。第一芯部区域412可以是纯pt。在其它实施方案中,第一芯部区域412由任何以下值或在以下值中任两个的范围内的pt制成:90、91、92、93、94、95、96、97、98、99和99.5原子%。第二芯部区域414可以是纯m
i
金属。在其它实施方案中,第二芯部区域414由任何以下值或在以下值中任两个的范围内的m
i
金属制成:90、91、92、93、94、95、96、97、98、99和99.5原子%。
[0056]
图8f描绘了负载在催化剂载体层378上的多元素催化剂结构360的示意性侧视图。多元素催化剂结构360包括区域416、第一芯部区域418和第二芯部区域420。区域416的形状通常为卵形。第一芯部区域418的形状通常为卵形。第二芯部区域420的形状通常为球形。区域416的一部分与催化剂载体层378直接接触。区域416被配置为负载第一和第二芯部区域418和420。区域416的一部分与第一芯部区域418界面接触。第一芯部区域418和第二芯部区域420的部分与彼此界面接触。区域416由m
ii
和m
iii
金属的基质形成。在所述区域416中m
ii
金
属与m
iii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。第一芯部区域418由pt和m
ii
金属的基质形成。在所述第一芯部区域418中pt与m
ii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。第二芯部区域420可以是纯m
i
金属。在其它实施方案中,第二芯部区域420由任何以下值或在以下值中任两个的范围内的m
i
金属制成:90、91、92、93、94、95、96、97、98、99和99.5原子%。
[0057]
图8g描绘了负载在催化剂载体层380上的多元素催化剂结构362的示意性侧视图。多元素催化剂结构362包括区域422、第一芯部区域424和第二芯部区域426。区域422的形状通常为球形。第一芯部区域424的形状通常为卵形。第二芯部区域426的形状通常为球形。区域422的一部分与催化剂载体层380直接接触。区域422被配置为负载第一和第二芯部区域424和426。区域422的一部分与第一芯部区域424界面接触。第一芯部区域424和第二芯部区域426的部分与彼此界面接触。区域422可以是纯m
ii
金属。在其它实施方案中,第二芯部区域426由任何以下值或在以下值中任两个的范围内的m
ii
金属制成:90、91、92、93、94、95、96、97、98、99和99.5原子%。第一芯部区域424由pt和m
ii
金属的基质形成。在所述第一芯部区域424中pt与m
ii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。第二芯部区域426由m
i
和m
ii
金属的基质形成。在所述第二芯部区域426中m
i
金属与m
ii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。
[0058]
图8h描绘了负载在催化剂载体层382上的多元素催化剂结构364的示意性侧视图。多元素催化剂结构364包括区域428、第一芯部区域430和第二芯部区域432。区域428的形状通常为卵形。第一芯部区域430的形状通常为卵形。第二芯部区域432的形状通常为球形。区域428的一部分与催化剂载体层382直接接触。区域428被配置为负载第一和第二芯部区域430和432。区域428的一部分与第一芯部区域430界面接触。第一芯部区域430和第二芯部区域432的部分与彼此界面接触。区域428由m
ii
和m
iii
金属的基质形成。在所述区域428中m
ii
金属与m
iii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。第一芯部区域430由pt和m
ii
金属的基质形成。在所述第一芯部区域430中pt与m
ii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。第二芯部区域432由m
i
和m
ii
金属的基质形成。在所述第二芯部区域432中m
i
金属与m
ii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。
[0059]
图8i描绘了负载在催化剂载体层384上的多元素催化剂结构366的示意性侧视图。多元素催化剂结构366包括区域434、第一芯部区域436和第二芯部区域438。区域434的形状通常为卵形。第一芯部区域436的形状通常为卵形。第二芯部区域438的形状通常为球形。区域434的一部分与催化剂载体层384直接接触。区域434被配置为负载第一和第二芯部区域436和438。区域434的一部分与第一芯部区域436界面接触。第一芯部区域436和第二芯部区域438的部分与彼此界面接触。区域434由m
ii
和m
iii
金属的基质形成。在所述区域434中m
ii
金属与m
iii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。第一芯部区域436可以是纯pt。在其它实施方案中,第一芯部区域436由任何以下值或在以下值中任两个的范围内的pt制成:90、91、92、93、94、95、96、97、98、99
和99.5原子%。第二芯部区域438由m
i
和m
ii
金属的基质形成。在所述第二芯部区域438中m
i
金属与m
ii
金属的原子比可以为任何以下值或者在以下值中任两个的范围内:0.5:1、0.75:1、1:1、1:0.75和1:0.5。
[0060]
关于图8a至图8i所示的多元素催化剂结构,在所述区域、第一芯部区域和第二芯部区域390中的材料的原子比可以是任何以下值或者在以下值中任两个的范围内:0.01:0.01:0.98、0.01:0.98:0.01、0.98:0.01:0.01和0.33:0.33:0.33。在某些实施方案中,所述第二芯部区域部分地包埋在所述第一芯部区域中,其中所述第二芯部区域的一部分表面暴露。在其它实施方案中,所述第二芯部区域完全地包埋在所述第一芯部区域中,其中所述第二芯部区域表面部分均没有暴露。
[0061]
在一个或多个实施方案中,使用扫描探针嵌段共聚物光刻(spbcl)来合成一个或多个实施方案的多元素催化剂结构。所述spbcl方法使用光刻法将聚合物圆顶限定为纳米反应器以合成所述多元素催化剂结构。
[0062]
尽管上面描述了示例性实施方案,但并不旨在使这些实施方案描述权利要求包括的所有可能形式。说明书中使用的措辞是描述性的措辞而不是限制性的,并且应当理解,在不脱离本公开的精神和范围的情况下可以进行各种改变。如前所述,各种实施方案的特征可以组合以形成可能没有明确描述或示出的本发明的其它实施方案。尽管可能已将各种实施方案描述为在一个或多个所需特性方面提供了优点或优于其它实施方案或现有技术的实施方式,但是本领域普通技术人员认识到可以折衷一个或多个特征或特性来实现所需的总体系统属性,这取决于特定的应用和实施。这些属性可以包括但不限于成本、强度、耐用性、寿命周期成本、市场性、外观、包装、尺寸、适用性、重量、可制造性、易于组装等。因此,对于任何实施方案描述为相比于其它实施方案或现有技术实施方式而言在一个或多个特征方面较不合意的程度而言,这些实施方案并不在本公开的范围之外,并且对于特定应用而言可能是合意的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1