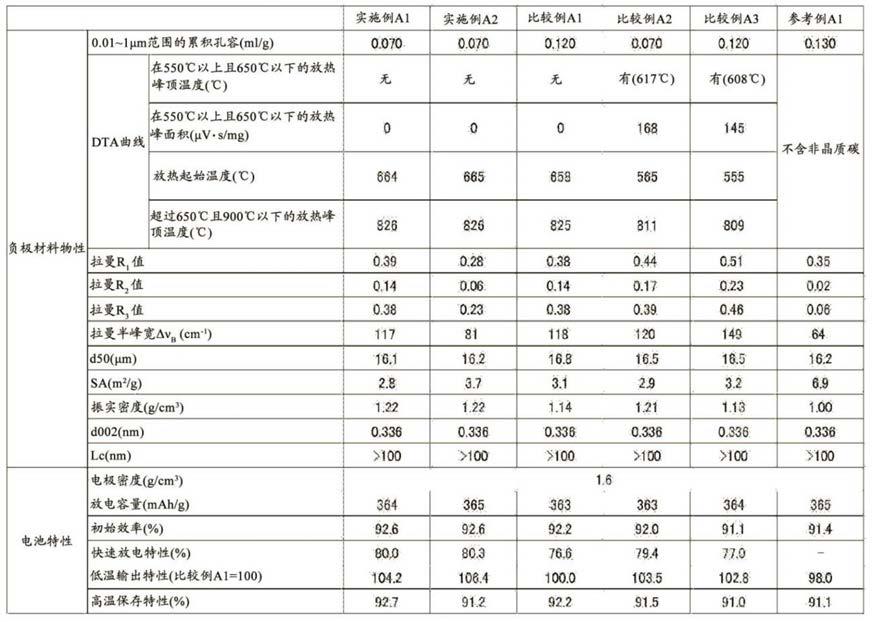
非水二次电池用负极材料原料、非水二次电池用负极材料、非水二次电池用负极及非水二次电池的制作方法
1.本发明涉及非水二次电池用负极材料原料、非水二次电池用负极材料、包含该非水二次电池用负极材料的非水二次电池用负极及非水二次电池。
背景技术:
2.近年来,伴随着电子设备的小型化,对高容量二次电池的需求不断提高。特别是,与镍镉电池及镍氢电池相比能量密度更高、快速充放电特性更优异的非水二次电池、尤其是锂离子二次电池备受关注。特别是,已开发了包含能够吸留和放出锂离子的正极及负极、以及溶解有lipf6、libf4等锂盐的非水电解液的非水锂二次电池,并已付诸实用。
3.作为该非水锂二次电池的负极材料,提出了各种材料,但出于高容量、放电电位的平坦性优异等理由,已被使用的是天然石墨、焦炭等经石墨化而得到的人造石墨、石墨化中间相沥青、石墨化碳纤维等石墨质的碳材料。另外,出于对于部分电解液比较稳定等理由,也使用了非晶质的碳材料。此外,还使用了在石墨粒子的表面包覆或附着有非晶质碳而兼备基于石墨的高容量且不可逆容量小这样的特性、以及基于非晶质碳的与电解液的稳定性优异这样的特性这两个特性的碳材料。
4.最近,在电动汽车等领域中,高容量、低温输入输出特性优异等特性受到重视,从这些观点出发,在上述的非水锂二次电池的负极材料中,使用了在石墨粒子的表面包覆或附着有非晶质碳的材料。在这样的材料中,专利文献1中公开了通过在特定条件下使非晶质碳包覆、同时对石墨进行各向同性加压处理而使快速充电特性得到改善的负极材料。另外,专利文献2中,对于包覆于石墨粒子的非晶质碳,公开了一种负极材料,其在原料中使用喹啉含量高的煤焦油沥青来控制存在于表面的硫元素及氮元素的原子浓度,由此成为高容量、且充放电特性得到了改善。另外,专利文献3中公开了一种负极材料,其通过具有特定的热特性的碳材料而使得加压载荷低、低温输出特性得到了改善。此外,专利文献4中公开了一种负极材料,其被特定的非晶质碳包覆、具有特定的孔容,由此使低温输入输出特性及高温保存特性得到了改善。
5.现有技术文献
6.专利文献
7.专利文献1:wo2011/052452
8.专利文献2:wo2012/157590
9.专利文献3:日本特开2017
‑
045574号公报
10.专利文献4:日本特开2018
‑
163868号公报
11.专利文献5:日本特开2017
‑
37711号公报
技术实现要素:
12.发明要解决的问题
13.根据本发明人等的研究可知,对于专利文献1所记载的非水二次电池用负极材料而言,起因于包覆石墨的非晶质碳的成分,其高温保存特性不充分。另外可知,对于专利文献2~4所记载的非水二次电池用负极材料而言,其快速充放电特性、及高温保存特性不充分。即,本发明的第1课题在于,提供一种非水二次电池用负极材料、以及包含该非水二次电池用负极材料的非水二次电池用负极及非水二次电池,所述非水二次电池用负极材料可以提供高容量、快速充放电特性优异、低温输入输出特性及高温保存特性的平衡优异的非水二次电池。
14.另外,根据本发明人等的研究可知,对于专利文献1~4所记载的非水二次电池用负极材料而言,会引起充放电循环后的极板膨胀。即,本发明的第2课题在于,提供一种非水二次电池用负极材料原料、非水二次电池用负极材料、以及包含其的非水二次电池用负极及非水二次电池,所述非水二次电池用负极材料能够提供高容量、快速充电特性优异、充放电循环后的极板膨胀小的非水二次电池。
15.根据本发明人等的研究可知,对于专利文献1所记载的非水二次电池用负极材料而言,其虽然为了控制微孔量而通过加压处理进行了改性、并通过包覆处理进行了调整,但单纯地增多非晶质碳物质量时,难以使粒子变硬而压制成高密度。另外可知,对于专利文献2~4所记载的非水二次电池用负极材料而言,虽然通过包覆非晶质碳物质而使快速充放电特性及高温保存特性得到了提高,但起因于非晶质碳物质,无法使粒子变硬而将电极压制成高密度。此外,专利文献5中考虑与非晶质碳物质量的压制性的关系而改善了容量、输出特性,但其所实施的负载、密度低,对于进一步的高密度化而言是不充分的。即,本发明的第3课题在于,提供一种非水二次电池用负极材料、以及包含该非水二次电池用负极材料的非水二次电池用负极及非水二次电池,所述非水二次电池用负极材料为了制作高容量的电池而用非晶质碳物质进行了包覆,其通过以更小的加压负载而压制成高密度,由此实现高容量、且极板膨胀小。
16.解决问题的方法
17.本发明人等为了解决上述的第1课题而进行深入研究的结果发现,通过将孔容控制为特定范围、且热特性满足特定的条件的非水二次电池用负极材料可以解决上述课题。即,本发明的第1方式的要点如以下所述。
18.[a1]一种非水二次电池用负极材料,其包含在表面的至少一部分具有非晶质碳物质的石墨,
[0019]
所述非水二次电池用负极材料中微孔径为0.01μm以上且1μm以下的范围的累积孔容为0.100ml/g以下,并且在由空气气流中的差示热分析法(dta)得到的dta曲线中,满足下述条件1)及2)中的至少一者,
[0020]
1)在550℃以上且650℃以下的温度范围不具有放热峰,
[0021]
2)在550℃以上且650℃以下的温度范围具有放热峰,并且该放热峰的面积超过0μv
·
s/mg且为90μv
·
s/mg以下。
[0022]
[a2]一种非水二次电池用负极材料,该非水二次电池用负极材料的由以下的式α计算出的拉曼r1值为0.15以上且1.00以下、拉曼半峰宽(δν
b
)为65cm
‑1以上且400cm
‑1以下,
[0023]
所述非水二次电池用负极材料中微孔径为0.01μm以上且1μm以下的范围的累积孔容为0.100ml/g以下,并且在由空气气流中的差示热分析法(dta)得到的dta曲线中,满足下
述条件1)及2)中的至少一者,
[0024]
1)在550℃以上且650℃以下的温度范围不具有放热峰,
[0025]
2)在550℃以上且650℃以下的温度范围具有放热峰,并且该放热峰的面积超过0μv
·
s/mg且为90μv
·
s/mg以下,
[0026]
式α:
[0027]
[拉曼r1值]=[拉曼光谱分析中1360cm
‑1附近的峰p
b
的强度i
b
]/[1580cm
‑1附近的峰p
a
的强度i
a
]。
[0028]
[a3]上述[a1]或[a2]所述的非水二次电池用负极材料,其中,在所述dta曲线中,满足下述条件3),
[0029]
3)在550℃以上且750℃以下的温度范围具有放热起始温度。
[0030]
[a4]上述[a1]~[a3]中任一项所述的非水二次电池用负极材料,其中,在所述dta曲线中,满足下述条件4),
[0031]
4)在超过650℃且为1000℃以下的温度范围具有放热峰。
[0032]
[a5]权利要求1~4中任一项所述的非水二次电池用负极材料,其中,该非水二次电池用负极材料的由以下的式β计算出的拉曼r2值为0.03以上且0.60以下、由以下的式γ计算出的拉曼r3值为0.10以上且1.00以下,
[0033]
式β:
[0034]
[拉曼r2值]=[拉曼光谱分析中1580cm
‑1附近的峰p
a
与1360cm
‑1附近的峰p
b
之间的谷(极小值)的强度i
c
]/[1580cm
‑1附近的峰p
a
的强度i
a
]
[0035]
式γ:
[0036]
[拉曼r3值]=[拉曼光谱分析中1580cm
‑1附近的峰p
a
与1360cm
‑1附近的峰p
b
之间的谷(极小值)的强度i
c
]/[1360cm
‑1附近的峰p
b
的强度i
b
]。
[0037]
[a6]上述[a1]~[a5]中任一项所述的非水二次电池用负极材料,其bet比表面积(sa)为0.5m2/g以上且10.0m2/g以下。
[0038]
[a7]上述[a1]~[a6]中任一项所述的非水二次电池用负极材料,其振实密度为0.60g/cm3以上且1.40g/cm3以下。
[0039]
[a8]上述[a1]~[a7]中任一项所述的非水二次电池用负极材料,其中,通过x射线衍射法求出的该非水二次电池用负极材料的面间距(d002)为0.340nm以下、且c轴方向的微晶尺寸(lc)为90nm以上。
[0040]
[a9]一种非水二次电池用负极,其具备:
[0041]
集电体、和
[0042]
形成在该集电体上的活性物质层,
[0043]
其中,该活性物质层含有上述[a1]~[a8]中任一项所述的非水二次电池用负极材料。
[0044]
[a10]一种非水二次电池,其是具备正极、负极及电解质的非水二次电池,其中,该负极为上述[a9]所述的非水二次电池用负极。
[0045]
本发明人等为了解决上述的第2课题而进行深入研究的结果发现,通过使用了将利用压汞法测定的退汞量/进汞量(b/a)控制为特定范围的石墨质原料的非水二次电池用负极材料,可以解决上述的第2课题。即,本发明的第2方式的要点如以下所述。
[0046]
[b1]一种非水二次电池用负极材料原料,其包含石墨,并且在将通过压汞法得到的进汞量设为a、将退汞量设为b时,下述式(1)为45%以上。
[0047]
式(1)b/a
×
100(%)
[0048]
[b2]上述[b1]所述的非水二次电池用负极材料原料,其中,所述进汞量a为0.001ml/g以上且0.5ml/g以下。
[0049]
[b3]上述[b1]或[b2]所述的非水二次电池用负极材料原料,其中,所述退汞量b为0.0005ml/g以上且0.5ml/g以下。
[0050]
[b4]上述[b1]~[b3]中任一项所述的非水二次电池用负极材料原料,其振实密度为0.7g/cm3以上且1.4g/cm3以下。
[0051]
[b5]一种非水二次电池用负极材料,其包含上述[1]~[4]中任一项所述的非水二次电池用负极材料原料。
[0052]
[b6]上述[b5]所述的非水二次电池用负极材料,其中,通过x射线衍射法得到的该非水二次电池用负极材料的面间距(d002)为0.34nm以下、且c轴方向的微晶尺寸(lc)为90nm以上。
[0053]
[b7]上述[b5]或[b6]所述的非水二次电池用负极材料,该非水二次电池用负极材料的由下述式α计算出的拉曼r1值为0.15以上且1.00以下。
[0054]
式α:
[0055]
[拉曼r1值]=[拉曼光谱分析中1360cm
‑1附近的峰p
b
的强度i
b
]/[1580cm
‑1附近的峰p
a
的强度i
a
]
[0056]
[b8]一种非水二次电池,其使用了上述[b5]~[b7]中任一项所述的负极材料。
[0057]
本发明人等为了解决上述的第2课题而进行了深入研究,结果发现可得到一种非水二次电池用负极材料、以及包含该非水二次电池用负极材料的非水二次电池用负极及非水二次电池,所述非水二次电池用负极材料通过控制单位覆盖率的粒子内的孔容、且控制粒子内空隙的峰,可以提供相对于粒子的硬度、压制性良好且极板膨胀小的非水二次电池,从而完成了本发明。
[0058]
[c1]一种非水二次电池用负极材料,其包含在表面的至少一部分具有非晶质碳物质或石墨质物质中的至少一者的石墨,
[0059]
所述非水二次电池用负极材料通过压汞法测定的微孔分布具有2个以上的峰,将微孔径为最小的峰与下一个峰的谷间的极小值以下的累积孔容设为y[ml/g]、将石墨表面的非晶质碳物质及石墨质物质的覆盖率设为x(%)时,满足下述式(1)及(2),所述微孔径为最小的峰的峰顶的微孔径为360nm以下。
[0060]
式(1):y>0.005
[0061]
式(2):y<
‑
0.006x+0.12。
[0062]
[c2]一种非水二次电池用负极材料,其包含在表面的至少一部分具有非晶质碳物质或石墨质物质中的至少一者的石墨,
[0063]
所述非水二次电池用负极材料通过压汞法测定的微孔分布具有2个以上的峰,将微孔径为最小的峰与下一个峰的谷间的极小值以下的累积孔容设为y[ml/g]、将石墨表面的非晶质碳物质及石墨质物质的覆盖率设为x(%)时,满足下述式(1)及(3)。
[0064]
式(1):y>0.005
[0065]
式(3):y<
‑
0.006x+0.079
[0066]
[c3]上述[c1]或[c2]所述的非水二次电池用负极材料,其中,所述石墨为天然石墨。
[0067]
[c4]上述[c1]~[c3]中任一项所述的非水二次电池用负极材料,其真密度为2.20g/cm3以上且小于2.262g/cm3。
[0068]
[c5]上述[c1]~[c4]中任一项所述的非水二次电池用负极材料,其振实密度为0.85g/cm3以上。
[0069]
[c6]上述[c1]~[c5]中任一项所述的非水二次电池用负极材料,其表面的至少一部分被非晶质碳物质覆盖。
[0070]
[c7]上述[c1]~[c6]中任一项所述的非水二次电池用负极材料,其含有球状石墨粒子,所述球状石墨粒子是将鳞片状石墨、鳞状石墨、及块状石墨进行造粒处理而成的。
[0071]
[c8]上述[c7]所述的非水二次电池用负极材料,其中,所述造粒处理是至少赋予冲击、压缩、摩擦、及剪切力中的任意的力学能的处理。
[0072]
[c9]上述[c7]或[c8]所述的非水二次电池用负极材料,其中,所述造粒处理是以下的处理:在壳体内具备高速旋转的旋转构件、且壳体内具有设置有多个叶片的转子的装置中,通过该转子进行高速旋转而对于被导入至内部的石墨赋予冲击、压缩、摩擦、及剪切力中的任意作用来进行造粒。
[0073]
[c10]一种非水二次电池用负极,其具备:集电体、和形成在该集电体上的活性物质层,该活性物质层含有上述[c1]~[c9]中任一项所述的负极材料。
[0074]
[c11]一种非水二次电池,其具备正极、负极及电解质,其中,该负极是上述[c10]所述的非水二次电池用负极。
[0075]
发明的效果
[0076]
根据本发明,通过第1方式,可提供一种非水二次电池用负极材料、以及包含该非水二次电池用负极材料的非水二次电池用负极及非水二次电池,所述非水二次电池用负极材料可以提供高容量、快速充放电特性优异、低温输入输出特性及高温保存特性的平衡优异的非水二次电池。
[0077]
根据本发明的第2方式,可提供一种非水二次电池用负极材料原料、非水二次电池用负极材料、以及包含其的非水二次电池用负极及非水二次电池,所述非水二次电池用负极材料原料及非水二次电池用负极材料可以提供高容量、快速充电特性优异、循环后的极板膨胀小的非水二次电池。
[0078]
根据本发明的第3方式,由于面向高容量的电池,相对于粒子的硬度能够以小的负载的压制而实现高密度、且极板膨胀小,由此可提供能够使非水二次电池高容量化的非水二次电池用负极材料、以及包含该非水二次电池用负极材料的非水二次电池用负极及非水二次电池。
附图说明
[0079]
图1是示出实施例a1、a2、比较例a1~a3及参考例a1中的高温保存特性与低温输出特性的关系的图表。
[0080]
图2是示出实施例a3、a4、比较例a4、a5中的高温保存特性与低温输出特性的关系
的图表。
[0081]
图3是示出实施例b1~b2及比较例b1~b3的退汞量/进汞量(b/a)与循环膨胀的关系的图表。
[0082]
图4是示出实施例c1~c3、比较例c1~c6中的覆盖率与基于压汞法得到的微孔分布的峰的极小值以下的微孔径的孔容的关系的图表。
[0083]
图5是示出实施例c1~c3、比较例c1~c6中的压制到1.65g/cm3所需的负载相对于pellet
‑
tap的关系的图表。
[0084]
图6是示出实施例c1和比较例c1的压汞法测定结果的图表。
具体实施方式
[0085]
以下,对本发明进行详细说明,但本发明并不限定于以下说明,可以在不脱离本发明主旨的范围内任意变形后实施。需要说明的是,在本发明中,使用“~”并在其前后夹着数值或物性值地表现的情况下,使用的是包含其前后的值的含义。
[0086]
<a.第1实施方式>
[0087]
〔a1.非水二次电池用负极材料〕
[0088]
本发明的一个实施方式涉及的非水二次电池用负极材料(以下有时简称为“负极材料”)在其表面的至少一部分包含具有非晶质碳物质的石墨,该非水二次电池用负极材料中微孔径为0.01μm以上且1μm以下的范围的累积孔容为0.100ml/g以下,并且在由空气气流中的差示热分析法(dta)得到的dta曲线中,满足下述条件1)及2)中的至少一者。
[0089]
1)在550℃以上且650℃以下的温度范围不具有放热峰;
[0090]
2)在550℃以上且650℃以下的温度范围具有放热峰,并且该放热峰的面积超过0μv
·
s/mg且为90μv
·
s/mg以下。
[0091]
本实施方式的非水二次电池用负极材料发挥以下效果:高容量,且快速充放电特性优异、低温输入输出特性及高温保存特性的平衡优异。本发明发挥这样的效果的理由虽然并不确定,但认为是以下理由。
[0092]
本实施方式的负极材料中,微孔径为0.01μm以上且1μm以下范围的累积孔容成为存在于负极材料粒子内部的孔容的指标。通过将其减小至特定的范围,在将电极压制到达到一定密度时,与以往的存在于粒子内部的孔容大的负极材料相比,能够相对地确保粒子间的孔容较大。其结果可认为,在充放电时li离子能够在电极内顺利地移动,从而快速充放电特性、低温输入输出特性优异。
[0093]
此外,一般来说,非晶质碳物质比石墨容易燃烧,因此存在与石墨相比在低温侧具有放热峰的倾向,特别是碳结构的发达程度不足的非晶质碳物质的情况下,该倾向表现得更显著。对于这样的碳结构的发达程度不足的非晶质碳物质而言,li离子无法在非晶质碳物质内部顺利地移动,因此快速充放电特性、低温输入输出特性降低,并且被充电后的li离子容易发生自放电、且在粒子表面容易引起与电解液的副反应,因此存在高温保存特性变差的倾向。另外,由于非晶质碳物质所具有的不可逆容量的影响、与电解液的副反应增加,存在电池的初始充放电效率降低、电池容量降低的倾向。
[0094]
另一方面,本实施方式的负极材料中,通过将非晶质碳物质的碳结构控制的足够发达、并具有特定的热特性,li离子可以在非晶质碳物质内部顺利地移动,因此显示出优异
的快速充放电特性、低温输入输出特性。另外,与此同时,还可以抑制被充电后的li离子的自放电、与电解液的副反应,因此可以推定会显示出优异的高温保存特性。另外,由于非晶质碳物质所具有的不可逆容量的影响、与电解液的副反应被抑制,因此存在电池的初始充放电效率提高、电池容量提高的倾向。
[0095]
另外,本发明的另一实施方式的负极材料为以下的负极材料:
[0096]
该非水二次电池用负极材料的由以下的式α计算出的拉曼r1值为0.20以上1.00以下、拉曼半峰宽(δν
b
)为65cm
‑1以上且400cm
‑1以下,该负极材料中微孔径为0.01μm以上且1μm以下范围的累积孔容为0.100ml/g以下,并且在由空气气流中的差示热分析法(dta)得到的dta曲线中,满足下述条件1)及2)中的至少一者。
[0097]
1)在550℃以上且650℃以下的温度范围不具有放热峰,
[0098]
2)在550℃以上且650℃以下的温度范围具有放热峰,并且该放热峰的面积超过0μv
·
s/mg且为90μv
·
s/mg以下,
[0099]
式α:
[0100]
[拉曼r1值]=[拉曼光谱分析中1360cm
‑1附近的峰p
b
的强度i
b
]/[1580cm
‑1附近的峰p
a
的强度i
a
]。
[0101]
本实施方式的非水二次电池用负极材料发挥以下效果:高容量,且快速充放电特性优异、低温输入输出特性及高温保存特性的平衡优异。本发明发挥这样的效果的理由虽然并不确定,但认为是以下理由。
[0102]
本实施方式的负极材料中,微孔径为0.01μm以上且1μm以下范围的累积孔容成为存在于负极材料粒子内部的孔容的指标。通过将其减小至特定的范围,在将电极压制到达到一定密度时,与以往的存在于粒子内部的孔容大的负极材料相比,能够相对地确保粒子间的孔容较大。其结果可认为,在充放电时li离子能够在电极内顺利地移动,从而快速充放电特性、低温输入输出特性优异。
[0103]
此外,拉曼r1值、拉曼半峰宽(δν
b
)成为负极材料粒子表面的石墨结晶性的指标。将它们增大至特定的范围表示的是降低负极材料粒子表面的石墨结晶性(提高非晶性)。可认为通过将它们增大至特定的范围,负极材料粒子表面的li离子的插入/脱离性变得良好,快速充放电特性、低温输入输出特性优异。该负极材料粒子表面的石墨结晶性例如可以通过使石墨结晶性低的碳物质添加附着于负极材料粒子表面的至少一部分来进行调节。
[0104]
一般来说,石墨结晶性低的碳物质比石墨容易燃烧,因此存在与石墨相比在低温侧具有放热峰的倾向,特别是碳结构的发达程度不足的碳物质的情况下,该倾向表现得更显著。对于这样的碳结构的发达程度不足的碳物质而言,li离子无法在碳物质内部顺利地移动,因此快速充放电特性、低温输入输出特性降低,并且被充电后的li离子容易发生自放电、且在粒子表面容易引起与电解液的副反应,因此存在高温保存特性变差的倾向。另外,由于碳结构的发达程度不足的碳物质所具有的不可逆容量的影响、与电解液的副反应增加,存在电池的初始充放电效率降低、电池容量降低的倾向。
[0105]
另一方面,本实施方式的负极材料中,通过将石墨结晶性低的碳物质的碳结构控制的足够发达、并具有特定的热特性,li离子可以在石墨结晶性低的碳物质内部顺利地移动,因此显示出优异的快速充放电特性、低温输入输出特性。另外,与此同时,还可以抑制被充电后的li离子的自放电、与电解液的副反应,因此可以推定会显示出优异的高温保存特
性。另外,由于石墨结晶性低的碳物质所具有的不可逆容量的影响、与电解液的副反应被抑制,因此存在电池的初始充放电效率提高、电池容量提高的倾向。
[0106]
[a1
‑
1.物性]
[0107]
本发明的一个实施方式的负极材料满足以下说明的孔容及热特性。另外,本发明的一个实施方式的负极材料满足以下说明的拉曼特性。另外,本发明的一个实施方式的负极材料优选满足以下的各物性。
[0108]
<a1
‑1‑
1.累积孔容>
[0109]
在本发明的一个实施方式的负极材料中,0.01μm以上且1μm以下范围中的累积孔容是使用压汞法(水银孔隙率法)测得的值,该累积孔容为0.100ml/g以下、优选为0.080ml/g以下、更优选为0.070ml/g以下、进一步优选为0.060ml/g以下、特别优选为0.050ml/g以下、最优选为0.030ml/g以下,另一方面,优选为0.001ml/g以上、更优选为0.002ml/g以上、进一步优选为0.005ml/g以上、特别优选为0.010ml/g以上、最优选为0.020ml/g以上。如果偏离上述范围,则在充放电时li离子不能在电极内顺利地移动,因此存在快速充放电特性、低温输入输出特性降低的倾向。
[0110]
作为用于上述汞孔隙率法的装置,可以使用水银孔隙率仪(autopore9520:micrometritics公司制造)。称量试样(碳材料)使其为0.2g左右的值,封入到粉末用容器中,在室温、真空下(50μmhg以下)进行10分钟脱气来实施前处理。接着,减压至4psia(约28kpa),向上述容器中导入水银,使压力从4psia(约28kpa)阶梯状地升压至40000psia(约280mpa),然后降压至25psia(约170kpa)。升压时的阶梯数设为80点以上,在各阶梯中保持10秒钟的平衡时间后,测定水银压入量。使用washburn式由这样得到的水银压入曲线计算出微孔分布。需要说明的是,以水银的表面张力(γ)为485dyne/cm、接触角(ψ)为140
°
来进行计算。平均微孔径定义为累积微孔体积(累积孔容)达到50%时的微孔径。
[0111]
<a1
‑1‑
2.热特性>
[0112]
对于本发明的一个实施方式的负极材料而言,在由空气气流中的差示热分析法(dta)得到的dta曲线中,满足以下的条件1)及2)中的至少一者。另外,在上述dta曲线中,优选满足条件3)、进一步优选满足条件4)。
[0113]
1)在550℃以上且650℃以下的温度范围不具有放热峰。
[0114]
2)在550℃以上且650℃以下的温度范围具有放热峰,并且该放热峰的面积超过0μv
·
s/mg且为90μv
·
s/mg以下。
[0115]
3)在550℃以上且750℃以下的温度范围具有放热起始温度。
[0116]
4)在超过650℃且为1000℃以下的温度范围具有放热峰。
[0117]
这里,上述各温度范围的放热峰是指,在它们的温度范围具有峰顶的放热峰。来源于非晶质碳的上述dta曲线的峰通常存在于550℃以上,满足条件1及2)中的至少一者意味着非晶质碳物质的碳结构足够发达,以热更稳定的状态存在。通过满足条件1)及2)中的至少一者,li离子能够在非晶质碳物质内部顺利地移动,因此显示出快速充放电特性、低温输入输出特性。另外,与此同时,可以抑制被充电了的li离子的自放电、与电解液的副反应,因此显示出优异的高温保存特性。其结果,快速充放电特性优异、低温输入输出特性及高温保存特性的平衡变得良好。需要说明的是,条件1)及2)中的“放热峰”是指放热峰的峰顶,条件2中的“放热峰的面积”是指放热峰整体的面积,而并不限定于550℃以上且650℃以下的温
度范围。
[0118]
本发明的一个实施方式的负极材料在由空气气流中的差示热分析法(dta)得到的dta曲线中在550℃以上且650℃以下的温度范围的放热峰的面积优选为90μv
·
s/mg以下、更优选为70μv
·
s/mg以下、进一步优选为50μv
·
s/mg以下、特别优选为30μv
·
s/mg以下、最优选为10μv
·
s/mg以下,理想的是0μv
·
s/mg。
[0119]
另外,在本发明的一个实施方式的负极材料中,除了满足条件1)及2)中的至少一者以外还满足条件3)意味着非晶质碳物质的碳结构更加发达、以热更稳定的状态存在。通过除了满足条件1)及2)中的至少一者以外还满足条件3),li离子能够在非晶质碳物质内部更顺利地移动。另外,与此同时,可以进一步抑制被充电了的li离子的自放电、与电解液的副反应,因此,快速充放电特性优异、低温输入输出特性及高温保存特性的平衡更加良好,因而优选。
[0120]
在本发明的一个实施方式的负极材料中,条件3)是由于非晶质碳物质的碳结构发达而在存在于dta曲线的峰的起始温度中显示优选范围的条件。在本发明的一个实施方式的负极材料中,由空气气流中的差示热分析法得到的dta曲线的放热起始温度优选为550℃以上、更优选为570℃以上、进一步优选为590℃以上、特别优选为600℃以上、最优选为620℃以上,另一方面,优选为750℃以下、更优选为720℃以下、进一步优选为700℃以下、特别优选为680℃以下、最优选为670℃以下。
[0121]
在本发明的一个实施方式的负极材料中进一步满足条件4)意味着控制了非晶质碳物质的碳结构不过于发达、li离子的插入/脱离位点不会过于减少。通过进一步满足条件4),具有适度的li离子的插入/脱离位点,快速充放电特性、低温输入输出特性变得良好,因而优选。需要说明的是,上述dta曲线中超过650℃且1000℃以下的温度范围的放热峰除了来源于非晶质碳物质的放热峰以外,还具有来源于石墨的放热峰,因此,呈现为这些放热峰的叠加。
[0122]
本发明的一个实施方式的负极材料优选在由空气气流中的差示热分析法(dta)得到的dta曲线中在特定的温度范围具有放热峰。作为该温度范围,优选为超过650℃、更优选为700℃以上、进一步优选为750℃以上、特别优选为800℃以上,另一方面,优选为1000℃以下、更优选为900℃以下、进一步优选为850℃以下、特别优选为830℃以下。
[0123]
需要说明的是,由空气气流中的差示热分析法得到的dta曲线可以使用hitachi high
‑
technologies公司制造的热重
‑
差热分析装置(tg
‑
dta)来进行测定。具体地,可以在氧化铝制试料容器中填充15mg的样品,以200ml/min的流量流通空气、以升温速度5℃/min从室温升温至1000℃的范围来进行测定。
[0124]
<a1
‑1‑
3.拉曼特性(拉曼r1值、拉曼半峰宽δν
b
(cm
‑1)、拉曼r2值、拉曼r3值)>
[0125]
(拉曼r1值)
[0126]
本说明书中的拉曼r1值如下述式α所示那样,定义为对负极材料通过拉曼分光法得到的拉曼光谱中测定1580cm
‑1附近的峰p
a
的强度i
a
与1360cm
‑1附近的峰p
b
的强度i
b
时的强度比(i
b
/i
a
)。需要说明的是,“1580cm
‑1附近”是指1580~1620cm
‑1的范围,“1360cm
‑1附近”是指1350~1370cm
‑1的范围。
[0127]
式α:
[0128]
[拉曼r1值]=[拉曼光谱分析中1360cm
‑1附近的峰p
b
的强度i
b
]/[1580cm
‑1附近的
峰p
a
的强度i
a
]
[0129]
本说明书中的拉曼半峰宽δν
b
(cm
‑1)被定义为对负极材料通过拉曼分光法得到的拉曼光谱中的1360cm
‑1附近的峰p
b
的半峰宽。需要说明的是,“1360cm
‑1附近”是指1350~1370cm
‑1的范围。
[0130]
本说明书中的拉曼r2值如下述式β所述那样,被定义为对负极材料通过拉曼分光法得到的拉曼光谱中测定1580cm
‑1附近的峰p
a
的强度i
a
、与1580cm
‑1附近的峰p
a
和1360cm
‑1附近的峰p
b
之间的谷(极小值)的强度i
c
时的强度比(i
c
/i
a
)。需要说明的是,“1580cm
‑1附近”是指1580~1620cm
‑1的范围,“1360cm
‑1附近”是指1350~1370cm
‑1的范围。
[0131]
式β:
[0132]
[拉曼r2值]=[拉曼光谱分析中1580cm
‑1附近的峰p
a
与1360cm
‑1附近的峰p
b
之间的谷(极小值)的强度i
c
]/[1580cm
‑1附近的峰p
a
的强度i
a
]
[0133]
本说明书中的拉曼r3值如下述式γ所述那样,被定义为对负极材料通过拉曼分光法得到的拉曼光谱中测定1360cm
‑1附近的峰p
b
的强度i
b
、与1580cm
‑1附近的峰p
a
和1360cm
‑1附近的峰p
b
之间的谷(极小值)的强度i
c
时的强度比(i
c
/i
b
)。需要说明的是,“1580cm
‑1附近”是指1580~1620cm
‑1的范围,“1360cm
‑1附近”是指1350~1370cm
‑1的范围。
[0134]
式γ:
[0135]
[拉曼r3值]=[拉曼光谱分析中1580cm
‑1附近的峰p
a
与1360cm
‑1附近的峰p
b
之间的谷(极小值)的强度i
c
]/[1360cm
‑1附近的峰p
b
的强度i
b
]
[0136]
本发明的一个实施方式的负极材料的拉曼r1值通常为0.15以上、优选为0.20以上、更优选为0.25以上、进一步优选为0.30以上、特别优选为0.32以上、最优选为0.35以上,另外,通常为1.00以下、优选为0.80以下、更优选为0.70以下、进一步优选为0.65以下、特别优选为0.50以下。
[0137]
本发明的一个实施方式的负极材料的拉曼半峰宽(δν
b
)通常为65cm
‑1以上、优选为70cm
‑1以上、更优选为80cm
‑1以上、进一步优选为85cm
‑1以上、特别优选为90cm
‑1以上、最优选为100cm
‑1以上,另外,通常为400cm
‑1以下、优选为300cm
‑1以下、更优选为250cm
‑1以下、进一步优选为200cm
‑1以下、特别优选为170cm
‑1以下、最优选为145cm
‑1以下。
[0138]
本发明的一个实施方式的负极材料的拉曼r2值优选为0.03以上、更优选为0.05以上、进一步优选为0.07以上、特别优选为0.09以上、最优选为0.11以上,另外,通常为0.60以下、优选为0.40以下、更优选为0.30以下、进一步优选为0.25以下、特别优选为0.22以下。
[0139]
本发明的第二实施方式的负极材料的拉曼r3值优选为0.10以上、更优选为0.15以上、进一步优选为0.20以上、特别优选为0.25以上、最优选为0.30以上,另外,通常为1.00以下、优选为0.80以下、更优选为0.70以下、进一步优选为0.60以下、特别优选为0.45以下。
[0140]
这些拉曼特性成为负极材料粒子表面的石墨结晶性的指标,它们的数值越大,表示负极材料粒子表面的石墨结晶性越是降低(非晶性提高)。拉曼r1值(i
b
/i
a
)不仅受石墨化度(石墨结构的完整性)的影响,而且还受到石墨结晶表面的边缘及结晶边界的比率的影响,拉曼r1值越大,越是存在石墨化度降低、边缘及结晶边界的比率变高的倾向。因此,不仅是粒子表面的一部分具有非晶质碳质的情况、而且在因石墨被割断地粉碎等而使边缘比率增大的情况下,拉曼r1值均存在增大的情况。另一方面,拉曼半峰宽(δν
b
)受石墨结晶表面的边缘及结晶边界的比率的影响比较小,存在更明显地反映石墨化度(石墨结构的完整性)
的影响的倾向。因此,因粒子表面的一部分具有非晶质碳质而存在拉曼半峰宽(δν
b
)变大的倾向,但在基于粉碎等而使边缘比率增大时,则存在变化较小的倾向。另外,1580cm
‑1附近的峰p
a
与1360cm
‑1附近的峰p
b
之间的谷(极小值)的强度i
c
被认为源自石墨结晶结构缺陷、非晶质碳结构中的sp3性,存在拉曼r2值(i
c
/i
a
)、拉曼r3值(i
c
/i
b
)更加反映石墨化度(局部的石墨结构的完整性)的影响的倾向。因此,拉曼r2值(i
c
/i
a
)、拉曼r3值(i
c
/i
b
)存在通过在粒子表面的一部分具有非晶质碳质而变大的倾向,但在基于粉碎等而使边缘比率增大时,则存在变化较小的倾向。
[0141]
上述拉曼特性的各参数值为上述下限值以上表示的是负极材料表面的非晶性为适宜的范围,li离子变得容易插入/脱离,由此存在低温输入输出特性提高的倾向。另一方面,上述拉曼特性的各参数值如果为上述上限值以下,则非晶性高的碳所具有的不可逆容量的影响的增大及与电解液的副反应的增加被抑制,可防止锂离子二次电池的初始充放电效率的降低及气体产生量的增加,存在电池容量提高的倾向。
[0142]
拉曼光谱可以用拉曼分光器来测定。具体来说,通过使测定对象粒子自然落下并填充在测定池(cell)内来进行试料填充,对测定池内照射氩离子激光,同时使测定池在与该激光垂直的面内旋转来进行测定。
[0143]
氩离子激光的波长:514.5nm
[0144]
试样上的激光功率:25mw
[0145]
分辨率:4cm
‑1[0146]
测定范围:1100cm
‑1~1730cm
‑1[0147]
峰强度测定、峰的半峰宽测定:本底(background)处理、平滑(smoothing)处理(基于单纯平均的卷积5点(
コンボリュション5ポイント
))
[0148]
<a1
‑1‑
4.bet比表面积(sa)>
[0149]
本发明的一个实施方式的负极材料的基于bet法的比表面积(sa)优选为0.5m2/g以上、更优选为1.0m2/g以上、进一步优选为1.5m2/g以上、特别优选为2.0m2/g以上,另一方面,优选为10.0m2/g以下、更优选为6.5m2/g以下、进一步优选为5.0m2/g以下、特别优选为4.0m2/g以下。如果sa为上述下限值以上,则可确保li离子进出的部位,存在锂离子二次电池的快速充放电特性、低温输入输出特性变得良好的倾向。另一方面,如果sa为上述上限值以下,则活性物质对于电解液的活性变得过于过剩,与电解液的副反应被抑制而防止电池的初始充放电效率的降低、气体产生量的增加,存在电池容量提高的倾向。
[0150]
需要说明的是,bet比表面积(sa)可以使用mountech公司制造的macsorb来进行测定。具体可以如下测定:对于试料在氮流通下于100℃进行了3小时预减压干燥后,使用冷却至液氮温度、且氮气相对于大气压的相对压力值准确调节为0.3的氮氦混合气体,通过采用气体流动法的氮吸附bet1点法来进行测定。
[0151]
<a1
‑1‑
5.振实密度>
[0152]
本发明的一个实施方式的负极材料的振实密度(g/cm3)优选为0.60g/cm3以上、更优选为0.80g/cm3以上、进一步优选为1.00g/cm3以上、特别优选为1.10g/cm3以上、最优选为1.15g以上,另一方面,优选为1.40g/cm3以下、更优选为1.35g/cm3以下、进一步优选为1.30g/cm3以下、特别优选为1.25g/cm3以下。如果振实密度为上述下限值以上,则在制作极板时的条痕产生等的工序性变得良好,负极材料层的填充性提高,因此,压延性良好、容易
形成高密度的负极片,高密度化成为可能,制成电极体时li离子迁移路径的弯曲度变小、且粒子间空隙的形状整齐,因此电解液的移动变得顺利,快速充放电特性提高,从上述观点出发是优选的,另外,如果为上述上限值以下,则粒子的表面及内部具有适度的空间,粒子不会变得过硬,电极压制性优异,并且低温输入输出特性、快速充放电特性优异,从上述观点出发是优选的。
[0153]
需要说明的是,振实密度使用粉体密度测定器tapdenser kyt
‑
3000(seishin enterprise株式会社制)进行测定。具体地,使试料落入20cc的振实容器中,在填充满容器之后,进行1000次冲程长度为10mm的振实,将此时的密度作为振实密度。
[0154]
<a1
‑1‑
6.面间距(d002)及微晶尺寸(lc)>
[0155]
关于本发明的一个实施方式的负极材料的结晶性(石墨化度),优选基于学振法的x射线广角衍射法求出的(002)面的面间距(d002)为0.340nm以下,另外,优选c轴方向的微晶尺寸(lc)为90nm以上。d002值更优选为0.338nm以下、进一步优选为0.337nm以下。另外,lc更优选为95nm以上、进一步优选为100nm以上。如果d002、lc为上述范围,则放电容量增大,因此从能够将电池高容量化的观点出发是优选的。
[0156]
<a1
‑1‑
7.圆度>
[0157]
本发明的一个实施方式的负极材料的通过流式粒子图像分析求出的圆度优选为0.88以上、更优选为0.90以上、进一步优选为0.91以上、特别优选为0.92以上、最优选为0.93以上。如果是这样的圆度高的负极材料,则li离子扩散的弯曲度下降而使粒子间空隙中的电解液移动变得顺利,从而可以提高快速充放电特性,因此优选。另一方面,该圆度的理论上限为1,因此通常小于1、优选为0.99以下、更优选为0.98以下、进一步优选为0.97以下。从确保所得到的负极材料彼此的接触性而提高循环特性的观点出发,优选为上述上限以下。
[0158]
需要说明的是,圆度是使用流式粒子图像分析装置(东亚医疗电子株式会社制fpia
‑
2000)、进行基于等效圆直径的粒径分布的测定并通过计算出平均圆度而求得。圆度用以下的式子来定义,圆度为1时成为理论上的真球。
[0159]
[圆度]=[具有与粒子投影形状相同面积的等效圆的周长]/[粒子投影形状的实际周长]
[0160]
在该圆度的测定中,作为分散介质,使用了离子交换水,作为表面活性剂,使用了聚氧乙烯(20)单月桂酸酯。等效圆直径是指具有与拍摄的粒子图像相同投影面积的圆(等效圆)的直径,圆度是指,以等效圆的周长为分子、以拍摄的粒子投影图像的周长为分母而得到的比率。将所测定的等效直径为10~40μm范围的粒子的圆度取平均,作为圆度。
[0161]
<a1
‑1‑
8.体积基准平均粒径(平均粒径d50)>
[0162]
本发明的一个实施方式的负极材料的体积基准平均粒径(也记作“平均粒径d50”)优选为1μm以上、更优选为3μm以上、进一步优选为4μm以上、特别优选为5μm以上,另外,优选为50μm以下、更优选为40μm以下、进一步优选为30μm以下、特别优选为25μm以下、最优选为20μm以下。如果d50的值为上述下限值以上,则存在容易防止不可逆容量的增加、初始电池容量的损失的倾向,另一方面,如果d50的值为上述上限值以下,则存在可防止浆料涂布中的条痕产生等工序不良情况的发生、快速充放电特性、低温输入输出特性提高的倾向。
[0163]
平均粒径(d50)如下定义:使0.01g复合粒子悬浮于作为表面活性剂的聚氧乙烯山
梨糖醇酐单月桂酸酯(作为例子,可以举出tween 20(注册商标))的0.2质量%水溶液10ml中,将其作为测定样品导入到市售的激光衍射/散射式粒度分布测定装置(例如horiba制造的la
‑
920)中,对测定样品以60w的输出功率照射28khz的超声波1分钟后,在上述测定装置中测定体积基准的中位径,将其定义为平均粒径d50。
[0164]
[a1
‑
2.制造方法]
[0165]
本发明的一个实施方式的非水二次电池用负极材料的制造方法只要是可以制造包含表面的至少一部分具有非晶质碳的石墨、或者满足特定的拉曼特性(拉曼r1值及拉曼半峰宽(δν
b
))、以及满足前述孔容和前述热特性的负极材料的方法即可,没有特别限制,例如,可以通过对实施了球形化处理的球状石墨进行加压处理,并将非晶质碳前体(非晶质碳的原料)混合、烧成来进行制造。加压处理可列举各向异性、各向同性的加压处理,但从将前述孔容和前述热特性控制在特定范围内的观点出发,优选各向同性加压处理。另外,作为加压处理的条件,没有特别限定,可以通过在50mpa以上且300mpa以下进行处理而将前述孔容和前述热特性控制在特定范围内。对于加压处理的条件而言,从将前述孔容和前述热特性控制在特定范围内的观点出发,优选为100mpa以上、更优选为120mpa以上、进一步优选为140mpa以上、特别优选为160mpa以上、最优选为180mpa以上,另外,优选为280mpa以下、更优选为260mpa以下、进一步优选为240mpa以下、特别优选为230mpa以下、最优选为220mpa以下。上述的制造方法中,具体地,作为在表面的至少一部分具有非晶质碳的石墨能够通过加压处理而调整为特定范围的孔容,并且为了适于碳结构的发达而使用灰分、金属杂质量、喹啉不溶成分(qi)等较低的非晶质碳前体,且在适度的烧成温度、非活性气氛下进行烧成,从而能够将负极材料的孔容和热特性调整为特定范围,因此优选。
[0166]
<a1
‑2‑
1.石墨>
[0167]
本发明的一个实施方式的负极材料包含石墨。用于制造本发明的一个实施方式的负极材料的石墨优选显示以下的种类、物性者。需要说明的是,关于石墨的物性,只要未对其测定条件及定义进行特别说明,则与前述的对于负极材料进行了说明的那些同样。
[0168]
对于石墨的种类而言,只要是能够吸留和放出锂离子即可,其种类没有特别限定,可以是天然石墨、人造石墨中的任意石墨。作为天然石墨,可以是鳞片状石墨、块状石墨、土壤石墨等的任意石墨,但优选杂质少的石墨,优选根据需要实施公知的纯化处理后使用。作为人造石墨,可列举:对煤焦油沥青、煤炭类重油、常压渣油、石油类重油、芳香烃、含氮环状化合物、含硫环状化合物、聚苯、聚氯乙烯、聚乙烯醇、聚丙烯腈、聚乙烯醇缩丁醛、天然高分子、聚苯硫醚、聚苯醚、糠醇树脂、苯酚酚醛树脂、酰亚胺树脂等有机物在通常为2500℃以上、且通常为3200℃以下的范围的温度进行烧成、石墨化而得到的人造石墨。此时,也可以将含硅化合物、含硼化合物等用作石墨化催化剂。
[0169]
对于石墨的结晶性(石墨化度)而言,其基于x射线广角衍射法的(002)面的面间距(d002)通常为0.335nm以上且小于0.340nm。另外,d002值优选为0.338nm以下、更优选为0.337nm以下、进一步优选为0.336nm以下。
[0170]
对于石墨的形状而言,从快速充放电特性的观点出发,特别优选为球状石墨(球状化石墨)。作为对石墨粒子进行球状化的方法,可以通过使用公知的技术实施球形化处理来制造被球形化了的石墨粒子。例如可列举使用反复对粒子赋予以冲击力为主体并且也包括粒子的相互作用在内的压缩、摩擦、剪切力等机械作用的装置来进行。具体来说,优选如下
的装置:在壳体内部具有设置了多个叶片的转子,并通过该转子高速旋转对导入到内部的碳材料赋予冲击、压缩、摩擦、剪切力等机械作用,由此来进行表面处理的装置。另外,优选具有通过使石墨粒子循环而反复赋予机械作用的机构的装置。作为具体的装置,可列举例如:混合系统(hybridization system)(株式会社奈良机械制作制)、kryptron(earthtechnica公司制)、cf磨(宇部兴产株式会社制)、机械熔融系统(hosokawa micron公司制)、theta composer(株式会社德寿工作所制)等。这些当中,优选株式会社奈良机械制作所制造的混合系统。例如在使用前述的装置进行处理的情况下,旋转的转子的圆周速度没有特别限制,优选设为30m/秒~100m/秒、更优选设为40m/秒~100m/秒、进一步优选设为50m/秒~100m/秒。另外,处理虽然能够仅使碳物质通过来进行,但优选使其在装置内循环或滞留30秒钟以上来进行处理,更优选使其在装置内循环或滞留1分钟以上来进行处理。另外,也可以使用球状石墨,该球状石墨是使用多个鳞片状或鳞状石墨、及被磨碎的石墨微粉等石墨和造粒剂,按照前述专利文献4中记载的方法进行造粒而得到的。
[0171]
(a1
‑2‑1‑
1.圆度)
[0172]
本发明的一个实施方式的负极材料的原料所使用的石墨的通过流式粒子图像分析求出的圆度优选为0.88以上、更优选为0.90以上、进一步优选为0.91以上、特别优选为0.92以上、最优选为0.93以上。如果是这样的圆度高的石墨,则使用该石墨制造的负极材料的li离子扩散的弯曲度下降而使粒子间空隙中的电解液移动变得顺利,从而可以提高快速充放电特性,因此优选。另一方面,该圆度的理论上限为1,因此通常小于1、优选为0.99以下、更优选为0.98以下、进一步优选为0.97以下。从确保所得到的负极材料彼此的接触性而提高循环特性的观点出发,优选为上述上限以下。
[0173]
圆度是使用流式粒子图像分析装置(东亚医疗电子株式会社制fpia
‑
2000)、进行基于等效圆直径的粒径分布的测定并通过计算出平均圆度而求得。圆度用以下的式子来定义,圆度为1时成为理论上的真球。
[0174]
[圆度]=[具有与粒子投影形状相同面积的等效圆的周长]/[粒子投影形状的实际周长]
[0175]
在该圆度的测定中,作为分散介质,使用了离子交换水,作为表面活性剂,使用了聚氧乙烯(20)单月桂酸酯。等效圆直径是指具有与拍摄的粒子图像相同投影面积的圆(等效圆)的直径,圆度是指,以等效圆的周长为分子、以拍摄的粒子投影图像的周长为分母而得到的比率。将所测定的等效直径为10~40μm范围的粒子的圆度取平均,作为圆度。
[0176]
(a1
‑2‑1‑
2.振实密度)
[0177]
石墨的振实密度优选为0.60g/cm3以上、更优选为0.70g/cm3以上、进一步优选为0.80g/cm3以上、特别优选为0.855/cm3以上、最优选为0.90g/cm3以上,另一方面,通常为1.40g/cm3以下、优选为1.30g/cm3以下、更优选为1.20g/cm3以下。
[0178]
(a1
‑2‑1‑
3.体积基准平均粒径)
[0179]
石墨的体积基准平均粒径(d50)没有特别限定,通常为1μm以上、优选为3μm以上、更优选为5μm以上,且通常为50μm以下、优选为40μm以下、更优选为30μm以下。
[0180]
(a1
‑2‑1‑
4.bet比表面积)
[0181]
石墨的bet比表面积没有特别限定,通常为1.0m2/g以上、优选为1.5m2/g以上、更优选为2.0m2/g以上、进一步优选为3.0m2/g以上、特别优选为4.5m2/g以上、最优选为5.0m2/g
以上,另外,通常为30.0m2/g以下、优选为20.0m2/g以下、更优选为10.0m2/g以下。
[0182]
(a1
‑2‑1‑
5.拉曼r1值)
[0183]
石墨的由下述式α表示的拉曼r1值没有特别限定,优选为0.10以上且1.00以下。另外,拉曼r值更优选为0.15以上、进一步优选为0.20以上、特别优选为0.25以上,另一方面,更优选为0.80以下、进一步优选为0.60以下。
[0184]
式α:
[0185]
拉曼r1值=(拉曼光谱分析中1360cm
‑1附近的峰p
b
的强度i
b
)/(1580cm
‑1附近的峰p
a
的强度i
a
)
[0186]
<a1
‑2‑
2.石墨的加压处理>
[0187]
本发明的一个实施方式的负极材料的制造优选包含对石墨进行加压处理的工序。作为石墨的加压处理的方法,只要是能够进行加压的方法就没有特别限制,可列举例如:将原料石墨加入到橡胶型等的容器中、以水作为加压介质的静水压各向同性加压处理、以及以空气等气体作为加压介质的基于气压的各向同性加压处理。另外,也可以将原料石墨填充至模具中以单向加压沿恒定方向进行加压处理。
[0188]
作为石墨的加压处理的加压介质的压力,优选为5~400mpa的范围、更优选为30~350mpa的范围、进一步优选为50~300mpa的范围。如果为上述下限值以上,则可以使孔容为更小的范围,另外,如果压力为上述上限值以下,则存在容易抑制所得到的负极材料的比表面积增大的倾向。
[0189]
<a1
‑2‑
3.石墨的非晶质碳复合化>
[0190]
本发明的一个实施方式的负极材料包含在表面的至少一部分具有非晶质碳的石墨。该非晶质碳复合化的处理可通过将石墨与非晶质碳前体(非晶质碳的原料)混合并烧成来进行。通过在适宜的条件下进行石墨的非晶质碳复合化,可满足前述的热特性、并且还容易将孔容控制为前述的范围。
[0191]
作为非晶质碳前体,没有特别限定,可列举:焦油及沥青、萘、蒽及其衍生物等芳香烃类、酚醛树脂、聚乙烯醇等热塑性高分子等有机物。这些有机物前体可以仅使用一种,也可以组合2种以上使用。这些当中,从碳结构容易发达这样的观点出发,优选焦油、沥青、芳香烃类。
[0192]
非晶质碳前体中所含的灰分相对于非晶质碳前体的总重量优选为1重量%以下、更优选为0.5重量%以下、进一步优选为0.1重量%以下,另外,灰分的下限通常为0.1重量ppm以上。如果非晶质碳前体中所含的灰分为上述范围内,则容易满足前述的热特性。
[0193]
在本说明书中,非晶质碳前体中所含的金属杂质量定义为:将相对于非晶质碳前体的总重量的fe、al、si、ca的总含量除以残碳率而得到的值。相对于非晶质碳前体的总重量优选为1000重量ppm以下、更优选为400重量ppm以下、进一步优选为100重量ppm以下、特别优选为50重量ppm以下,且通常为0.1重量ppm以上。如果非晶质碳前体中所含的金属杂质量为上述范围内,则容易满足前述的热特性。
[0194]
非晶质碳前体中所含的喹啉不溶成分(qi)相对于非晶质碳前体的总重量优选为5重量%以下、更优选为2重量%以下、进一步优选为1重量%以下、特别优选为0.5重量%以下、最优选为0.1重量%以下。如果非晶质碳前体中所含的喹啉不溶成分(qi)为上述范围内,则容易满足前述的热特性。
[0195]
将石墨与非晶质碳前体混合后,进行烧成。进行烧成时的温度优选为950℃以上、更优选为1000℃以上、进一步优选为1050℃以上、特别优选为1100℃以上、最优选为1150℃以上,另一方面,优选为2000℃以下、更优选为1800℃以下、进一步优选为1600℃以下、特别优选为1500℃以下。另外,进行烧成时的时间优选为0.5小时以上、更优选为1小时以上,另一方面,优选为1000小时以下、更优选为500小时以下、进一步优选为100小时以下。如果进行烧成时的温度、时间为上述范围,则容易满足前述的热特性。
[0196]
进行烧成时的气氛优选为非活性气氛下。具体可列举:通过使氮、氩这样的非活性气体流通来降低氧浓度的方法、通过减压处理将氧排出至体系外并用氮、氩来恢复压力的方法、通过将焦炭粉等牺牲材料填充至制品的周围来降低炉内气氛中所含的氧的方法。可以通过非活性气体的流通量、流通时间、减压处理的程度、牺牲材料的填充条件等来控制体系内的氧浓度。氧浓度(体积浓度)优选为3%以下、更优选为1%以下、进一步优选为1000ppm以下、特别优选为500ppm以下、最优选为100ppm以下。如果超过上述范围,则非晶质碳物质的碳结构的发达受到阻碍,存在难以满足前述的热特性的倾向。
[0197]
在石墨中混合非晶质碳前体时的非晶质碳前体的混合比率应该基于目标的复合粒子的组成适宜选择,对于相对于石墨的非晶质碳前体的量而言,以作为其残炭物(非晶质碳)的重量比(〔[非晶质碳的重量]/[石墨的重量]〕
×
100)计,通常为0.01%以上、优选为0.1%以上、更优选为0.5%以上、进一步优选为1%以上、特别优选为2%以上、最优选为3%以上,另一方面,优选为30%以下、更优选为20%以下、进一步优选为15%以下、特别优选为10%以下。如果该重量比为上述范围,则为高容量、且li离子容易插入/脱离,因此低温输入输出特性、快速充放电特性及循环特性优异,从这点来看是优选的。
[0198]
<a1
‑2‑
4.其它处理>
[0199]
为了制造本发明的一个实施方式的负极材料,可以对通过前述的制造方法得到的复合粒子另行进行粉碎处理。
[0200]
作为粉碎处理所使用的粗粉碎机,可列举颚式破碎机、冲击式压碎机、锥形压碎机等,作为中间粉碎机,可列举辊式压碎机、锤磨机等,作为微粉碎机,可列举球磨、振动磨、销棒粉碎机、搅拌磨、喷射磨等。这些当中,从粉碎时间短、处理速度的观点出发,优选球磨、振动磨。
[0201]
粉碎速度根据装置的种类、大小适宜设定,例如,球磨的情况下,通常为50rpm以上、优选为100rpm以上、更优选为150rpm以上、进一步优选为200rpm以上,另外,通常为2500rpm以下、优选为2300rpm以下、更优选为2000rpm以下。如果速度过快,则存在控制粒径变得困难的倾向,如果速度过慢,则存在处理速度变慢的倾向。
[0202]
粉碎时间通常为30秒钟以上、优选为1分钟以上、更优选为1分30秒钟以上、进一步优选为2分钟以上,另外,通常为3小时以下、优选为2.5小时以下、更优选为2小时以下。如果粉碎时间过短,则存在粒径控制变得困难的倾向,如果粉碎时间过长,则存在生产性下降的倾向。
[0203]
振动磨的情况下,粉碎速度通常为50rpm以上、优选为100rpm以上、更优选为150rpm以上、进一步优选为200rpm以上,另外,通常为2500rpm以下、优选为2300rpm以下、更优选为2000rpm以下。如果速度过快,则存在粒径的控制变得困难的倾向,如果速度过慢,则存在处理速度变慢的倾向。
[0204]
粉碎时间通常为30秒钟以上、优选为1分钟以上、更优选为1分30秒钟以上、进一步优选为2分钟以上,另外,通常为3小时以下、优选为2.5小时以下、更优选为2小时以下。如果粉碎时间过短,则存在粒径控制变得困难的倾向,如果粉碎时间过长,则存在生产性下降的倾向。
[0205]
为了制造本发明的一个实施方式的负极材料,可以对通过前述的制造方法得到的复合粒子进行粒径的分级处理。作为分级处理条件,可以使用开孔通常为53μm以下、优选为45μm以下、更优选为38μm以下的装置来实施。
[0206]
作为用于分级处理的装置,没有特别限制,例如,在干式筛分的情况下,可以使用旋转式筛、摇动式筛、转动式筛、振动式筛等,在干式气流式分级的情况下,可以使用重力式分级机、惯性力式分级机、离心力式分级机(分粒器、旋风分离器)等,在湿式筛分的情况下,可以使用机械式湿式分级机、水力分级机、沉降分级机、离心式湿式分级机等。
[0207]
〔a2.非水二次电池用负极〕
[0208]
本发明的一个实施方式的非水二次电池用负极(以下,有时称为“本发明的负极”)具备:集电体、以及形成在该集电体上的活性物质层,该活性物质层含有上述的本发明的一个实施方式的负极材料。
[0209]
使用上述负极材料制作负极时,利用水性或有机类介质将在负极材料中配合有粘结树脂的材料制成浆料,并根据需要而向其中加入增稠剂后涂布在集电体、进行干燥即可。
[0210]
作为粘结树脂,优选使用相对于非水电解液稳定、且非水溶性的树脂。例如可使用:丁苯橡胶、异戊二烯橡胶及乙丙橡胶等橡胶状高分子;聚乙烯、聚丙烯、聚对苯二甲酸乙二醇酯、聚酰亚胺、聚丙烯酸、及芳香族聚酰胺等合成树脂;苯乙烯
‑
丁二烯
‑
苯乙烯嵌段共聚物或其加氢产物、苯乙烯
‑
乙烯
‑
丁二烯、苯乙烯共聚物、苯乙烯
‑
异戊二烯及苯乙烯嵌段共聚物以及其氢化物等热塑性弹性体;间规立构
‑
1,2
‑
聚丁二烯、乙烯
‑
乙酸乙烯酯共聚物、及乙烯与碳原子数3~12的α
‑
烯烃的共聚物等软质树脂状高分子;四氟乙烯
‑
乙烯共聚物、聚偏氟乙烯、聚五氟丙烯及聚六氟丙烯等氟化高分子等。作为有机类介质,可使用例如n
‑
甲基吡咯烷酮及二甲基甲酰胺。
[0211]
相对于负极材料100重量份,通常使用0.1重量份以上、优选使用0.2重量份以上的粘结树脂。通过使粘结树脂的使用量相对于负极材料100重量份为0.1重量份以上,负极材料相互间的粘结力、负极材料与集电体的粘结力变得充分,可以防止由于从负极剥离负极材料而引起的电池容量的减少及循环特性的变差。
[0212]
另外,粘结树脂的使用量相对于负极材料100重量份优选为10重量份以下、更优选为7重量份以下。通过使粘结树脂的使用量相对于负极材料100重量份为10重量份以下,可以防止负极容量的减少、且防止锂离子等碱金属离子相对于负极材料的出入受到阻碍等问题。
[0213]
作为添加到浆料中的增稠剂,可列举例如:羧甲基纤维素、甲基纤维素、羟乙基纤维素及羟丙基纤维素等水溶性纤维素类、聚乙烯醇及聚乙二醇等。这些当中,优选羧甲基纤维素。优选以相对于负极材料100重量份通常为0.1~10重量份、特别是0.2~7重量份来使用增稠剂。
[0214]
作为负极集电体,使用目前已知可用于该用途的例如铜、铜合金、不锈钢、镍、钛及碳等即可。集电体的形状通常为片状,还优选使用其表面带有凹凸的材料、网状物及冲孔金
属等。
[0215]
在集电体上涂布负极材料和粘结树脂的浆料并进行干燥之后,优选进行加压而使形成在集电体上的活性物质层的密度增大、从而使负极活性物质层的每单位体积的电池容量增大。活性物质层的密度优选在1.2~1.8g/cm3的范围、更优选为1.3~1.6g/cm3。通过使活性物质层的密度为上述下限值以上,可以防止伴随着电极厚度的增大而带来的电池容量的降低。另外,通过使活性物质层的密度为上述上限值以下,伴随电极内的粒子间空隙减少而导致空隙中所保持的电解液量减少,可以防止锂离子等碱金属离子的移动性变小而使快速充放电特性降低。
[0216]
〔a3.非水二次电池〕
[0217]
本发明的一个实施方式的非水二次电池是具备正极、负极、以及电解质的非水二次电池,其中,作为负极,使用的是本发明的一个实施方式的负极。特别是,本发明的一个实施方式的非水二次电池中使用的正极及负极通常优选为能够吸留和放出li离子的正极及负极,本发明的一个实施方式的非水二次电池优选为锂离子二次电池。
[0218]
本发明的一个实施方式的非水二次电池除了使用了上述的本发明的一个实施方式的负极以外,可以按照常规方法来制造。特别是,本发明的一个实施方式的非水二次电池优选将[负极的容量]/[正极的容量]的值设计成1.01~1.5、更优选设计成1.2~1.4。
[0219]
[a3
‑
1.正极]
[0220]
作为本发明的一个实施方式的非水二次电池的成为正极的活性物质的正极材料,例如使用基本组成以licoo2表示的锂钴复合氧化物、以linio2表示的锂镍复合氧化物、以limno2及limn2o4表示的锂锰复合氧化物等锂过渡金属复合氧化物、二氧化锰等过渡金属氧化物、以及这些复合氧化物的混合物等即可。进一步,可使用tis2、fes2、nb3s4、mo3s4、cos2、v2o5、cro3、v3o3、feo2、geo2及lini
0.33
mn
0.33
co
0.33
o2、lifepo4等。
[0221]
可以通过将在上述正极材料中配合有粘结树脂的材料利用适当的溶剂进行浆料化并涂布在集电体上并进行干燥而制作正极。需要说明的是,在浆料中,优选含有乙炔黑及科琴黑等导电材料。另外,也可以根据需要而含有增稠剂。需要说明的是,作为粘结材料及增稠剂,可以使用其用途公知的那些,例如可以使用作为用于负极的制造而示例出的那些。
[0222]
相对于正极材料100重量份,导电材料的配合量优选为0.5重量份~20重量份、更优选为1重量份~15重量份。另外,相对于正极材料100重量份,增稠剂的配合量优选为0.2重量份~10重量份、更优选为0.5重量份~7重量份。此外,就相对于正极材料100重量份的粘结树脂的配合量而言,在用水将粘结树脂浆料化的情况下,优选为0.2重量份~10重量份、更优选为0.5重量份~7重量份,另一方面,在用n
‑
甲基吡咯烷酮等溶解粘结树脂的有机溶剂将粘结树脂浆料化的情况下,优选为0.5重量份~20重量份、更优选为1~15重量份。
[0223]
作为正极集电体,可列举例如:铝、钛、锆、铪、铌及钽等、以及它们的合金。这些当中,优选铝、钛及钽、及其合金,最优选为铝及其合金。
[0224]
[a3
‑
2.电解液]
[0225]
电解液可使用在以往周知的非水溶剂中溶解各种锂盐而成的溶液。
[0226]
作为非水溶剂,可使用例如碳酸亚乙酯、氟代碳酸亚乙酯、碳酸亚丙酯、碳酸亚丁酯及碳酸亚乙烯酯等环状碳酸酯、碳酸二甲酯、碳酸甲乙酯及碳酸二乙酯等链状碳酸酯、γ
‑
丁内酯等环状酯、冠醚、2
‑
甲基四氢呋喃、四氢呋喃、1,2
‑
二甲基四氢呋喃及1,3
‑
二氧戊
环等环状醚、1,2
‑
二甲氧基乙烷等链状醚等。通常可以将这些溶剂中的2种以上混合使用。这些当中,优选使用环状碳酸酯和链状碳酸酯,或者在其中进一步混合其它溶剂后使用。
[0227]
也可以在电解液中添加碳酸亚乙烯酯、乙烯基碳酸亚乙酯、琥珀酸酐、马来酸酐、丙磺酸内酯及二乙基砜等化合物、二氟磷酸锂这样的二氟磷酸盐等。进一步,还可以添加二苯基醚及环己基苯等过充电防止剂。
[0228]
作为溶解于非水溶剂的电解质,可列举例如:liclo4、lipf6、libf4、licf3so3、lin(cf3so2)2、lin(cf3cf2so2)2、lin(cf3so2)(c4f9so2)及lic(cf3so2)3等。电解液中的电解质的浓度通常为0.5mol/l~2mol/l、优选为0.6mol/l~1.5mol/l。
[0229]
[a3
‑
3.隔板]
[0230]
本发明的一个实施方式的非水二次电池中优选使用隔板,且该隔板介于正极和负极之间。作为这样的隔板,优选使用聚乙烯、聚丙烯等聚烯烃的多孔性片、或无纺布。
[0231]
<b.第2实施方式>
[0232]
以下,对本发明进行详细说明,但本发明并不限定于以下说明,可以在不脱离本发明主旨的范围内任意变形后实施。需要说明的是,在本发明中,使用“~”并在其前后夹着数值或物性值地表现的情况下,使用的是包含其前后的值的含义。
[0233]
〔b1.非水二次电池用负极材料原料〕
[0234]
本发明的一个实施方式的非水二次电池用负极材料原料(以下,有时称为“本发明的负极材料原料”)包含石墨,并且在将通过压汞法测定的进汞量(ml/g)设为a、将退汞量(ml/g)设为b时,退汞量/进汞量(b/a)
×
100为45%以上。
[0235]
退汞量/进汞量(b/a)是负极材料原料粒子内结构的复杂程度的指标,粒子内结构复杂关系到粒子内的残留应力的大小,而粒子内的残留应力大则有可能会关系到伴随着使用了该原料的负极材料的充放电循环的粒子膨胀的产生容易程度。因此,退汞量/进汞量(b/a)
×
100通常为45%以上、更优选为50%以上、进一步优选为55%以上、特别优选为60%以上、最优选为63%以上。作为退汞量/进汞量(b/a)
×
100的上限,没有特别限定,粒子内结构的复杂程度越是降低(即,粒子内结构变得简单),越是存在粒子的球形度降低的倾向,如果粒子的球形度降低,则使用了采用该原料的负极材料的负极中的锂离子扩散性降低,对于快速充电性能等负载特性的不良影响可能会变得显著,因此上述退汞量/进汞量(b/a)
×
100优选为95%以下、更优选为90%以下、进一步优选为85%以下、最优选为80%以下。
[0236]
作为进汞量(ml/g)a,没有特别限定,a小则意味着粒子内微孔量少,如果粒子内微孔量少,则有可能会关系到使用了该原料的负极材料的输入输出特性等负载特性的降低。因此,进汞量(ml/g)a优选为0.001ml/g以上、更优选为0.003ml/g以上、进一步优选为0.005ml/g以上、特别优选为0.01ml/g以上、最优选为0.03ml/g以上。a大则意味着粒子内微孔量多,粒子内微孔量多有可能会关系到使用了该原料的负极材料的松密度的降低而引起的浆料制作效率、电极干燥效率的降低,因此,作为进汞量(ml/g)a的上限,优选为1.0ml/g以下、更优选为0.8ml/g以下、进一步优选为0.6ml/g以下、特别优选为0.4ml/g以下、最优选为0.3ml/g以下。
[0237]
作为退汞量(ml/g)b,没有特别限定,b小则反映的是粒子内微孔量少、或者粒子内结构复杂,前者有可能会关系到使用了该原料的负极材料的输入输出特性等负载特性的降低,后者反映出粒子内的残留应力大,而粒子内的残留应力大有可能会关系到担心伴随着
使用了该原料的负极材料的充放电循环的粒子膨胀的产生。因此,退汞量(ml/g)b优选为0.001ml/g以上、更优选为0.005ml/g以上、进一步优选为0.010ml/g以上、特别优选为0.040ml/g以上、最优选为0.065ml/g以上。b大则意味着刚刚测定b之前的进汞量(ml/g)a大,而a过大则存在前述的担心,因此,作为退汞量(ml/g)b的上限,优选为1.0ml/g以下、更优选为0.6ml/g以下、进一步优选为0.4ml/g以下、特别优选为0.2ml/g以下、最优选为0.1ml/g以下。
[0238]
上述这样的负极材料原料包含于负极材料中时,可以抑制伴随着二次电池的充放电的循环后的极板膨胀(循环膨胀),因此优选。
[0239]
本实施方式的非水二次电池用负极材料原料及非水二次电池用负极材料可发挥提供高容量、循环膨胀的抑制优异、且快速充电性能优异的非水二次电池这样的效果。本发明发挥这样的效果的理由虽然尚不确定,但认为是以下的理由。
[0240]
在本实施方式的负极材料原料中,退汞量/进汞量(b/a)成为粒子内结构的复杂程度的指标,b/a值越小,意味着粒子内结构越复杂。可认为,粒子内结构复杂关系到粒子内的残留应力的大小,而粒子内的残留应力大则有可能会关系到伴随着使用了该负极材料原料的负极材料的充放电循环的粒子膨胀的产生容易程度。通过使b/a
×
100的值为45%以上,认为可将粒子内结构的复杂程度抑制在一定程度以下,从而抑制粒子内的残留应力,由此,能够抑制伴随着使用了该负极材料原料的负极材料的充放电循环的粒子膨胀,负极的循环膨胀的抑制优异。
[0241]
此外,通过将b/a
×
100的值保持在63%以上,负极的循环膨胀的抑制优异,而且还可以提高快速充电性能。认为这是由于,负极中的锂离子扩散被促进,负极中的锂离子浓度梯度变缓,在快速充电这样的物质扩散为速率决定这样的使用条件下,浓度过电压被抑制。
[0242]
[b1
‑
1.物性]
[0243]
本发明的一个实施方式的负极材料原料满足以下说明的(b/a)
×
100。
[0244]
<b1
‑1‑
1.进汞量及退汞量>
[0245]
对于本发明的负极材料原料而言,在将通过压汞法测得的进汞量(ml/g)设为a、将退汞量(ml/g)设为b时,退汞量/进汞量(b/a)
×
100为45%以上。
[0246]
作为上述汞孔隙率法用装置,可以使用水银孔隙率仪(autopore 9520:micrometritics公司制造)。称量试样(负极材料原料)使其为0.2g左右的值,封入到粉末用容器中,在室温、真空下(50μmhg以下)进行10分钟脱气来实施前处理。接着,减压至4psia(约28kpa),向上述容器中导入水银,使压力从4psia(约28kpa)阶梯状地升压至40000psia(约280mpa),然后降压至25psia(约170kpa)。升压时的阶梯数设为42点以上、降压时的阶梯数设为39点以上,在各阶梯中保持10秒钟的平衡时间后,测定水银压入量。使用washburn式由这样得到的水银压入曲线计算出微孔分布。需要说明的是,以水银的表面张力(γ)为485dyne/cm、接触角(ψ)为140
°
来进行计算。平均微孔径定义为累积微孔体积达到50%时的微孔径。由这样得到的汞压入行为及汞退出行为,如下所述地计算出进汞量(a)和退汞量(b)。
[0247]
进汞量(a)=压入侧0.01μm以上且0.6μm以下的累积孔容(ml/g)
[0248]
退汞量(b)=直至退出侧25psia(约170kpa)的退汞量(ml/g)
[0249]
另外,本发明的一个实施方式的负极材料原料优选满足以下的各物性。
[0250]
<b1
‑1‑
2.圆度>
[0251]
本发明的一个实施方式的负极材料原料的通过流式粒子图像分析求出的圆度优选为0.88以上、更优选为0.90以上、进一步优选为0.91以上、特别优选为0.92以上、最优选为0.93以上。如果是这样的圆度高的石磨,则使用其制造的负极材料的li离子扩散的弯曲度下降而使粒子间空隙中的电解液移动变得顺利,从而可以提高快速充放电特性,因此优选。另一方面,该圆度的理论上限为1,因此通常小于1、优选为0.99以下、更优选为0.98以下、进一步优选为0.97以下。从确保所得到的负极材料彼此的接触性而提高循环特性的观点出发,优选为上述上限以下。
[0252]
需要说明的是,圆度是使用流式粒子图像分析装置(东亚医疗电子株式会社制fpia
‑
2000)、进行基于等效圆直径的粒径分布的测定并通过计算出平均圆度而求得。圆度用以下的式子来定义,圆度为1时成为理论上的真球。
[0253]
[圆度]=[具有与粒子投影形状相同面积的等效圆的周长]/[粒子投影形状的实际周长]
[0254]
在该圆度的测定中,作为分散介质,使用了离子交换水,作为表面活性剂,使用了聚氧乙烯(20)单月桂酸酯。等效圆直径是指具有与拍摄的粒子图像相同投影面积的圆(等效圆)的直径,圆度是指,以等效圆的周长为分子、以拍摄的粒子投影图像的周长为分母而得到的比率。将测得的等效直径为10~40μm范围的粒子的圆度取平均,作为圆度。
[0255]
<b1
‑1‑
3.振实密度>
[0256]
负极材料原料的振实密度优选为0.60g/cm3以上、更优选为0.70g/cm3以上、进一步优选为0.80g/cm3以上、特别优选为0.855/cm3以上、最优选为0.90g/cm3以上,另一方面,通常为1.40g/cm3以下、优选为1.30g/cm3以下、更优选为1.20g/cm3以下。
[0257]
需要说明的是,振实密度使用粉体密度测定器tapdenser kyt
‑
3000(seishin enterprise株式会社制)进行测定。具体地,使试料落入20cc的振实容器中,在填充满容器之后,进行1000次冲程长度为10mm的振实,将此时的密度作为振实密度。
[0258]
<b1
‑1‑
4.体积基准平均粒径>
[0259]
负极材料原料的体积基准平均粒径(d50)没有特别限定,通常为1μm以上、优选为3μm以上、更优选为5μm以上、进一步优选为8μm以上、特别优选为10μm以上,且通常为50μm以下、优选为40μm以下、更优选为30μm以下、进一步优选为20μm以下、特别优选为16μm以下、最优选为14μm以下。
[0260]
平均粒径(d50)如下定义:使0.01g复合粒子悬浮于作为表面活性剂的聚氧乙烯山梨糖醇酐单月桂酸酯(作为例子,可以举出tween 20(注册商标))的0.2质量%水溶液10ml中,将其作为测定样品导入到市售的激光衍射/散射式粒度分布测定装置(例如horiba制造的la
‑
920)中,对测定样品以60w的输出功率照射28khz的超声波1分钟后,在上述测定装置中测定体积基准的中位径,将其定义为平均粒径d50。
[0261]
<b1
‑1‑
5.bet比表面积>
[0262]
负极材料原料的bet比表面积没有特别限定,通常为1.0m2/g以上、优选为1.5m2/g以上、更优选为2.0m2/g以上、进一步优选为3.0m2/g以上、特别优选为4.5m2/g以上、最优选为5.0m2/g以上,另外,通常为30.0m2/g以下、优选为25.0m2/g以下、更优选为20.0m2/g以下、进一步优选为16.0m2/g以下、更进一步优选为13.0m2/g以下、再进一步优选为12.0m2/g以
下。
[0263]
需要说明的是,bet比表面积(sa)可以使用mountech公司制造的macsorb来进行测定。具体可以如下测定:对于试料在氮流通下于100℃进行了3小时预减压干燥后,使用冷却至液氮温度、且氮气相对于大气压的相对压力值准确调节为0.3的氮氦混合气体,通过采用气体流动法的氮吸附bet1点法来进行测定。
[0264]
〔b2.非水二次电池用负极材料〕
[0265]
本发明的非水二次电池用负极材料包含上述的本发明的非水二次电池用负极材料原料。
[0266]
[b2
‑
1.物性]
[0267]
本发明的一个实施方式的负极材料优选满足以下的各物性。
[0268]
(拉曼r1值)
[0269]
本说明书中的拉曼r1值如下述式α所示那样,定义为对负极材料通过拉曼分光法得到的拉曼光谱中测定1580cm
‑1附近的峰p
a
的强度i
a
与1360cm
‑1附近的峰p
b
的强度i
b
时的强度比(i
b
/i
a
)。需要说明的是,“1580cm
‑1附近”是指1580~1620cm
‑1的范围,“1360cm
‑1附近”是指1350~1370cm
‑1的范围。
[0270]
负极材料原料的下述式α表示的拉曼r1值没有特别限定,优选为0.10以上且1.00以下。另外,拉曼r值更优选为0.15以上、进一步优选为0.20以上、特别优选为0.25以上,另一方面,更优选为0.80以下、进一步优选为0.60以下。
[0271]
式α:
[0272]
拉曼r1值=(拉曼光谱分析中1360cm
‑1附近的峰p
b
的强度i
b
)/(1580cm
‑1附近的峰p
a
的强度i
a
)
[0273]
<b2
‑1‑
1.负极材料的拉曼特性(拉曼r1值、拉曼半峰宽δν
b
(cm
‑1)、拉曼r2值、拉曼r3值)>
[0274]
(拉曼r1值)
[0275]
如上述那样,本说明书中的拉曼r1值如下述式α所示那样,定义为对负极材料通过拉曼分光法得到的拉曼光谱中测定1580cm
‑1附近的峰p
a
的强度i
a
与1360cm
‑1附近的峰p
b
的强度i
b
时的强度比(i
b
/i
a
)。需要说明的是,“1580cm
‑1附近”是指1580~1620cm
‑1的范围,“1360cm
‑1附近”是指1350~1370cm
‑1的范围。
[0276]
式α:
[0277]
[拉曼r1值]=[拉曼光谱分析中1360cm
‑1附近的峰p
b
的强度i
b
]/[1580cm
‑1附近的峰p
a
的强度i
a
]
[0278]
本说明书中的拉曼半峰宽δν
b
(cm
‑1)被定义为对负极材料通过拉曼分光法得到的拉曼光谱中的1360cm
‑1附近的峰p
b
的半峰宽。需要说明的是,“1360cm
‑1附近”是指1350~1370cm
‑1的范围。
[0279]
本说明书中的拉曼r2值如下述式β所述那样,被定义为对负极材料通过拉曼分光法得到的拉曼光谱中测定1580cm
‑1附近的峰p
a
的强度i
a
、与1580cm
‑1附近的峰p
a
和1360cm
‑1附近的峰p
b
之间的谷(极小值)的强度i
c
时的强度比(i
c
/i
a
)。需要说明的是,“1580cm
‑1附近”是指1580~1620cm
‑1的范围,“1360cm
‑1附近”是指1350~1370cm
‑1的范围。
[0280]
式β:
[0281]
[拉曼r2值]=[拉曼光谱分析中1580cm
‑1附近的峰p
a
与1360cm
‑1附近的峰p
b
之间的谷(极小值)的强度i
c
]/[1580cm
‑1附近的峰p
a
的强度i
a
]
[0282]
本说明书中的拉曼r3值如下述式γ所述那样,被定义为对负极材料通过拉曼分光法得到的拉曼光谱中测定1360cm
‑1附近的峰p
b
的强度i
b
、与1580cm
‑1附近的峰p
a
和1360cm
‑1附近的峰p
b
之间的谷(极小值)的强度i
c
时的强度比(i
c
/i
b
)。需要说明的是,“1580cm
‑1附近”是指1580~1620cm
‑1的范围,“1360cm
‑1附近”是指1350~1370cm
‑1的范围。
[0283]
式γ:
[0284]
[拉曼r3值]=[拉曼光谱分析中1580cm
‑1附近的峰p
a
与1360cm
‑1附近的峰p
b
之间的谷(极小值)的强度i
c
]/[1360cm
‑1附近的峰p
b
的强度i
b
]
[0285]
本发明的一个实施方式的负极材料的拉曼r1值通常为0.15以上、优选为0.20以上、更优选为0.25以上、进一步优选为0.30以上、特别优选为0.32以上、最优选为0.35以上,另外,通常为1.00以下、优选为0.80以下、更优选为0.70以下、进一步优选为0.65以下、特别优选为0.50以下。
[0286]
本发明的一个实施方式的负极材料的拉曼半峰宽(δν
b
)通常为65cm
‑1以上、优选为70cm
‑1以上、更优选为80cm
‑1以上、进一步优选为85cm
‑1以上、特别优选为90cm
‑1以上、最优选为100cm
‑1以上,另外,通常为400cm
‑1以下、优选为300cm
‑1以下、更优选为250cm
‑1以下、进一步优选为200cm
‑1以下、特别优选为170cm
‑1以下、最优选为145cm
‑1以下。
[0287]
本发明的一个实施方式的负极材料的拉曼r2值优选为0.03以上、更优选为0.05以上、进一步优选为0.07以上、特别优选为0.09以上、最优选为0.11以上,另外,通常为0.60以下、优选为0.40以下、更优选为0.30以下、进一步优选为0.25以下、特别优选为0.22以下。
[0288]
这些拉曼特性成为负极材料表面的石墨结晶性的指标,它们的数值越大,表示负极材料表面的石墨结晶性越是降低(非晶性提高)。拉曼r1值(i
b
/i
a
)不仅受石墨化度(石墨结构的完整性)的影响,而且还受到石墨结晶表面的边缘及结晶边界的比率的影响,拉曼r1值越大,越是存在石墨化度降低、边缘及结晶边界的比率变高的倾向。因此,不仅是负极材料表面的一部分具有非晶质碳质的情况、而且在因石墨被割断地粉碎等而使边缘比率增大的情况下,拉曼r1值均存在增大的情况。另一方面,拉曼半峰宽(δν
b
)受石墨结晶表面的边缘及结晶边界的比率的影响比较小,存在更明显地反映石墨化度(石墨结构的完整性)的影响的倾向。因此,因负极材料表面的一部分具有非晶质碳质而存在拉曼半峰宽(δν
b
)变大的倾向,但在基于粉碎等而使边缘比率增大时,则存在变化较小的倾向。另外,1580cm
‑1附近的峰p
a
与1360cm
‑1附近的峰p
b
之间的谷(极小值)的强度i
c
被认为源自石墨结晶结构缺陷、非晶质碳结构中的sp3性,存在拉曼r2值(i
c
/i
a
)、拉曼r3值(i
c
/i
b
)更加反映石墨化度(局部的石墨结构的完整性)的影响的倾向。因此,拉曼r2值(i
c
/i
a
)、拉曼r3值(i
c
/i
b
)存在通过在负极材料表面的一部分具有非晶质碳质而变大的倾向,但在基于粉碎等而使边缘比率增大时,则存在变化较小的倾向。
[0289]
上述拉曼特性的各参数值为上述下限值以上表示的是负极材料表面的非晶性为适宜的范围,li离子变得容易插入/脱离,由此存在低温输入输出特性提高的倾向。另一方面,上述拉曼特性的各参数值如果为上述上限值以下,则非晶性高的碳所具有的不可逆容量的影响的增大及与电解液的副反应的增加被抑制,可防止锂离子二次电池的初始充放电效率的降低及气体产生量的增加,存在电池容量提高的倾向。
[0290]
拉曼光谱可以用拉曼分光器来测定。具体来说,通过使测定对象粒子自然落下并填充在测定池(cell)内来进行试料填充,对测定池内照射氩离子激光,同时使测定池在与该激光垂直的面内旋转来进行测定。
[0291]
氩离子激光的波长:514.5nm
[0292]
试样上的激光功率:25mw
[0293]
分辨率:4cm
‑1[0294]
测定范围:1100cm
‑1~1730cm
‑1[0295]
峰强度测定、峰的半峰宽测定:本底(background)处理、平滑(smoothing)处理(基于单纯平均的卷积5点(
コンボリュション5ポイント
))
[0296]
<b2
‑1‑
2.bet比表面积(sa)>
[0297]
本发明的一个实施方式的负极材料的基于bet法的比表面积(sa)优选为0.5m2/g以上、更优选为1.0m2/g以上、进一步优选为1.2m2/g以上、特别优选为1.5m2/g以上、最优选为1.8m2/g以上,另一方面,优选为10.0m2/g以下、更优选为6.5m2/g以下、进一步优选为5.0m2/g以下、特别优选为4.0m2/g以下、最优选为3.0m2/g以下。如果sa为上述下限值以上,则可确保li离子进出的部位,存在锂离子二次电池的快速充放电特性变得良好的倾向。另一方面,如果sa为上述上限值以下,则活性物质对于电解液的活性变得过于过剩,与电解液的副反应被抑制而防止电池的初始充放电效率的降低、气体产生量的增加,存在电池容量提高的倾向。bet比表面积(sa)通过与负极材料原料中所说明的方法同样的方法来进行测定。
[0298]
<b2
‑1‑
3.振实密度>
[0299]
本发明的一个实施方式的负极材料的振实密度(g/cm3)优选为0.60g/cm3以上、更优选为0.80g/cm3以上、进一步优选为1.00g/cm3以上、特别优选为1.10g/cm3以上、最优选为1.15g以上,另一方面,优选为1.40g/cm3以下、更优选为1.35g/cm3以下、进一步优选为1.30g/cm3以下、特别优选为1.24g/cm3以下。如果振实密度为上述下限值以上,则在制作极板化时的条痕产生等的工序性变得良好,负极材料层的填充性提高,因此,压延性良好、容易形成高密度的负极片,高密度化成为可能,制成电极体时li离子迁移路径的弯曲度变小、且粒子间空隙的形状整齐,因此电解液的移动变得顺利,快速充放电特性提高,从上述观点出发是优选的,另外,如果为上述上限值以下,则粒子的表面及内部具有适度的空间,粒子不会变得过硬,电极压制性优异,并且快速充放电特性优异,从上述观点出发是优选的。振实密度通过与负极材料原料中所说明的方法同样的方法来进行测定。
[0300]
<b2
‑1‑
4.面间距(d002)及微晶尺寸(lc)>
[0301]
关于本发明的一个实施方式的负极材料的结晶性(石墨化度),优选基于学振法的x射线广角衍射法求出的(002)面的面间距(d002)为0.340nm以下,另外,优选c轴方向的微晶尺寸(lc)为90nm以上。d002值更优选为0.338nm以下、进一步优选为0.337nm以下。另外,lc更优选为95nm以上、进一步优选为100nm以上。如果d002、lc为上述范围,则放电容量增大,因此从能够将电池高容量化的观点出发是优选的。
[0302]
<b2
‑1‑
5.圆度>
[0303]
本发明的一个实施方式的负极材料的通过流式粒子图像分析求出的圆度优选为0.88以上、更优选为0.90以上、进一步优选为0.91以上、特别优选为0.92以上、最优选为
0.93以上。如果是这样的圆度高的负极材料,则li离子扩散的弯曲度下降而使粒子间空隙中的电解液移动变得顺利,从而可以提高快速充放电特性,因此优选。另一方面,该圆度的理论上限为1,因此通常小于1、优选为0.99以下、更优选为0.98以下、进一步优选为0.97以下。从确保所得到的负极材料彼此的接触性而提高循环特性的观点出发,优选为上述上限以下。圆度通过与负极材料原料中所说明的方法同样的方法来求出。
[0304]
<b2
‑1‑
6.体积基准平均粒径(平均粒径d50)>
[0305]
本发明的一个实施方式的负极材料的体积基准平均粒径(也记作“平均粒径d50”)优选为1μm以上、更优选为3μm以上、进一步优选为4μm以上、特别优选为5μm以上、最优选为8μm以上,另外,优选为50μm以下、更优选为40μm以下、进一步优选为30μm以下、特别优选为20μm以下、最优选为14μm以下。如果d50的值为上述下限值以上,则存在容易防止不可逆容量的增加、初始电池容量的损失的倾向,另一方面,如果d50的值为上述上限值以下,则存在可防止浆料涂布中的条痕产生等工序不良情况的发生、快速充放电特性、低温输入输出特性提高的倾向。体积基准平均粒径通过与负极材料原料中所说明的方法同样的方法来求出。
[0306]
[b3.制造方法]
[0307]
本发明的一个实施方式的非水二次电池用负极材料原料的制造方法只要是可以制造包含石墨、且在将通过压汞法测定的进汞量(ml/g)设为a、将退汞量(ml/g)设为b时满足退汞量/进汞量(b/a)
×
100为45%以上的负极材料原料的方法即可,没有特别限制,例如,可以通过对实施了球形化处理的球状石墨进行加压处理来制造。加压处理可列举各向异性、各向同性的加压处理,但从将退汞量/进汞量(b/a)控制在特定范围内的观点出发,优选各向同性加压处理。另外,作为加压处理的条件,没有特别限定,可以通过在50mpa以上且300mpa以下进行处理而将退汞量/进汞量(b/a)控制在特定范围内。对于加压处理的条件而言,从将退汞量/进汞量(b/a)控制在特定范围内的观点出发,优选为100mpa以上、更优选为120mpa以上、进一步优选为140mpa以上、特别优选为160mpa以上、最优选为180mpa以上,另外,优选为280mpa以下、更优选为260mpa以下、进一步优选为240mpa以下、特别优选为230mpa以下、最优选为220mpa以下。对于上述的制造方法而言,具体地,例如如下所述:将天然石墨粉碎,在粉碎后的天然石墨中混合造粒剂来进行球形化处理,进一步通过热处理而除去造粒剂,从而得到球形化石墨,对所得到的球形化石墨进行加压处理,将所得到的成型物进行破碎、分级处理,从而可以制造退汞量/进汞量(b/a)被控制在特定范围的非水二次电池用负极材料原料。
[0308]
另外,本发明的一个实施方式的非水二次电池用负极材料的制造方法没有特别限定,可以通过将本发明的一个实施方式的非水二次电池用负极材料原料与非晶质碳前体(非晶质碳的原料)混合并进行烧成来制造。具体地,优选使用灰分、金属杂质量、喹啉不溶成分(qi)等较低的非晶质碳前体,并在适度的烧成温度、非活性气氛下进行烧成,使得制成球形化石磨可通过加压处理来调整孔容、从而实现特定范围的退汞量/进汞量,并且适于碳结构的发达。
[0309]
<b3
‑
1.石墨>
[0310]
本发明的一个实施方式的负极材料原料包含石墨。用于制造本发明的一个实施方式的负极材料原料的石墨优选显示以下的种类、物性者。需要说明的是,关于石墨的物性,只要未对其测定条件及定义进行特别说明,则与前述的对于负极材料原料进行了说明的那
些同样。
[0311]
对于石墨的种类而言,只要是能够吸留和放出锂离子即可,其种类没有特别限定,可以是天然石墨、人造石墨中的任意石墨。作为天然石墨,可以是鳞片状石墨、块状石墨、土壤石墨等的任意石墨,但优选杂质少的石墨,优选根据需要实施公知的纯化处理后使用。作为人造石墨,可列举:对煤焦油沥青、煤炭类重油、常压渣油、石油类重油、芳香烃、含氮环状化合物、含硫环状化合物、聚苯、聚氯乙烯、聚乙烯醇、聚丙烯腈、聚乙烯醇缩丁醛、天然高分子、聚苯硫醚、聚苯醚、糠醇树脂、苯酚酚醛树脂、酰亚胺树脂等有机物在通常为2500℃以上、且通常为3200℃以下的范围的温度进行烧成、石墨化而得到的人造石墨。此时,也可以将含硅化合物、含硼化合物等用作石墨化催化剂。
[0312]
对于石墨的结晶性(石墨化度)而言,其基于x射线广角衍射法的(002)面的面间距(d002)通常为0.335nm以上且小于0.340nm。另外,d002值优选为0.338nm以下、更优选为0.337nm以下、进一步优选为0.336nm以下。
[0313]
对于石墨的形状而言,从快速充放电特性的观点出发,特别优选为球状石墨(球状化石墨)。作为对石墨粒子进行球状化的方法,可以通过使用公知的技术实施球形化处理来制造被球形化了的石墨粒子。作为球形化处理,例如可列举使用反复对粒子赋予以冲击力为主体并且也包括粒子的相互作用在内的压缩、摩擦、剪切力等机械作用的装置来进行。作为用于球形化的装置,具体来说,优选如下的装置:在壳体内部具有设置了多个叶片的转子,并通过该转子高速旋转对导入到内部的碳材料赋予冲击、压缩、摩擦、剪切力等机械作用,由此来进行表面处理的装置。另外,优选具有通过使石墨粒子循环而反复赋予机械作用的机构的装置。作为具体的装置,可列举例如:混合系统(hybridizationsystem)(株式会社奈良机械制作制)、kryptron
[0314]
(earthtechnica公司制)、cf磨(宇部兴产株式会社制)、机械熔融系统(hosokawa micron公司制)、theta composer(株式会社德寿工作所制)等。这些当中,优选株式会社奈良机械制作所制造的混合系统。例如在使用前述的装置进行处理的情况下,旋转的转子的圆周速度没有特别限制,优选设为30m/秒~100m/秒、更优选设为40m/秒~100m/秒、进一步优选设为50m/秒~100m/秒。另外,处理虽然能够仅使碳物质通过来进行,但优选使其在装置内循环或滞留30秒钟以上来进行处理,更优选使其在装置内循环或滞留1分钟以上来进行处理。另外,也可以使用球状石墨,该球状石墨是使用多个鳞片状或鳞状石墨、及被磨碎的石墨微粉等石墨和造粒剂,按照前述专利文献4中记载的方法进行造粒而得到的。
[0315]
(圆度)
[0316]
用作本发明的一个实施方式的负极材料原料的石墨的通过流式粒子图像分析求出的圆度优选为0.88以上、更优选为0.90以上、进一步优选为0.91以上、特别优选为0.92以上、最优选为0.93以上。如果是这样的圆度高的石墨,则使用该石墨制造的负极材料的li离子扩散的弯曲度下降而使粒子间空隙中的电解液移动变得顺利,从而可以提高快速充放电特性,因此优选。另一方面,该圆度的理论上限为1,因此通常小于1、优选为0.99以下、更优选为0.98以下、进一步优选为0.97以下。从确保所得到的负极材料彼此的接触性而提高循环特性的观点出发,优选为上述上限以下。
[0317]
(振实密度)
[0318]
石墨的振实密度优选为0.60g/cm3以上、更优选为0.70g/cm3以上、进一步优选为
0.80g/cm3以上、特别优选为0.855/cm3以上、最优选为0.90g/cm3以上,另一方面,通常为1.40g/cm3以下、优选为1.30g/cm3以下、更优选为1.20g/cm3以下。
[0319]
(体积基准平均粒径)
[0320]
石墨的体积基准平均粒径(d50)没有特别限定,通常为1μm以上、优选为3μm以上、更优选为5μm以上,且通常为50μm以下、优选为40μm以下、更优选为30μm以下。
[0321]
(bet比表面积)
[0322]
石墨的bet比表面积没有特别限定,通常为1.0m2/g以上、优选为1.5m2/g以上、更优选为2.0m2/g以上、进一步优选为3.0m2/g以上、特别优选为4.5m2/g以上、最优选为5.0m2/g以上,另外,通常为30.0m2/g以下、优选为25.0m2/g以下、更优选为20.0m2/g以下。
[0323]
(拉曼r1值)
[0324]
石墨的由下述式α表示的拉曼r1值没有特别限定,优选为0.10以上且1.00以下。另外,拉曼r值更优选为0.15以上、进一步优选为0.20以上、特别优选为0.25以上,另一方面,更优选为0.80以下、进一步优选为0.60以下。
[0325]
式α:
[0326]
拉曼r1值=(拉曼光谱分析中1360cm
‑1附近的峰p
b
的强度i
b
)/(1580cm
‑1附近的峰p
a
的强度i
a
)
[0327]
<b3
‑
2.石墨的加压处理>
[0328]
本发明的一个实施方式的负极材料原料的制造优选包含对石墨进行加压处理的工序。作为石墨的加压处理的方法,只要是能够进行加压的方法就没有特别限制,可列举例如:将原料石墨加入到橡胶型等的容器中、以水作为加压介质的静水压各向同性加压处理、以及以空气等气体作为加压介质的基于气压的各向同性加压处理。另外,也可以将原料石墨填充至模具中以单向加压沿恒定方向进行加压处理。
[0329]
作为石墨的加压处理的加压介质的压力,优选为5~400mpa的范围、更优选为30~350mpa的范围、进一步优选为50~300mpa的范围。如果为上述下限值以上,则可以使孔容为更小的范围,另外,如果压力为上述上限值以下,则存在容易抑制所得到的负极材料的比表面积增大的倾向。
[0330]
<b3
‑
3.负极材料原料的非晶质碳复合化>
[0331]
本发明的一个实施方式的负极材料优选包含在表面的至少一部分具有非晶质碳的石墨。该非晶质碳复合化的处理通过将包含上述的石墨的负极材料原料与非晶质碳前体(非晶质碳的原料)混合并进行烧成来进行。通过在适当的条件下进行包含石墨的负极材料原料的非晶质碳复合化,容易将累积孔容控制在前述的范围。
[0332]
作为非晶质碳前体,没有特别限定,可列举:焦油及沥青、萘、蒽及其衍生物等芳香烃类、酚醛树脂、聚乙烯醇等热塑性高分子等有机物。这些有机物前体可以仅使用一种,也可以组合2种以上使用。这些当中,从碳结构容易发达这样的观点出发,优选焦油、沥青、芳香烃类。
[0333]
非晶质碳前体中所含的灰分相对于非晶质碳前体的总重量优选为1重量%以下、更优选为0.5重量%以下、进一步优选为0.1重量%以下,另外,灰分的下限通常为0.1重量ppm以上。如果非晶质碳前体中所含的灰分为上述范围内,则热特性变得良好。
[0334]
本发明中的非晶质碳前体中所含的金属杂质量定义为:将相对于非晶质碳前体的
总重量的fe、al、si、ca的总含量除以残碳率而得到的值。相对于非晶质碳前体的总重量优选为1000重量ppm以下、更优选为400重量ppm以下、进一步优选为100重量ppm以下、特别优选为50重量ppm以下,且通常为0.1重量ppm以上。如果非晶质碳前体中所含的金属杂质量为上述范围内,则热特性变得良好。
[0335]
非晶质碳前体中所含的喹啉不溶成分(qi)相对于非晶质碳前体的总重量优选为5重量%以下、更优选为2重量%以下、进一步优选为1重量%以下、特别优选为0.5重量%以下、最优选为0.1重量%以下。如果非晶质碳前体中所含的喹啉不溶成分(qi)为上述范围内,则热特性变得良好。
[0336]
将石墨与非晶质碳前体混合后,进行烧成。进行烧成时的温度优选为950℃以上、更优选为1000℃以上、进一步优选为1050℃以上、特别优选为1100℃以上、最优选为1150℃以上,另一方面,优选为2000℃以下、更优选为1800℃以下、进一步优选为1600℃以下、特别优选为1500℃以下。另外,进行烧成时的时间优选为0.5小时以上、更优选为1小时以上,另一方面,优选为1000小时以下、更优选为500小时以下、进一步优选为100小时以下。如果进行烧成时的温度、时间为上述范围,则热特性变得良好。
[0337]
进行烧成时的气氛优选为非活性气氛下。具体可列举:通过使氮、氩这样的非活性气体流通来降低氧浓度的方法、通过减压处理将氧排出至体系外并用氮、氩来恢复压力的方法、通过将焦炭粉等牺牲材料填充至制品的周围来降低炉内气氛中所含的氧的方法。可以通过非活性气体的流通量、流通时间、减压处理的程度、牺牲材料的填充条件等来控制体系内的氧浓度。氧浓度(体积浓度)优选为3%以下、更优选为1%以下、进一步优选为1000ppm以下、特别优选为500ppm以下、最优选为100ppm以下。如果超过上述范围,则非晶质碳物质的碳结构的发达受到阻碍,存在难以得到理想的热特性的倾向。
[0338]
在石墨中混合非晶质碳前体时的非晶质碳前体的混合比率应该基于目标的复合粒子的组成适宜选择,对于相对于石墨的非晶质碳前体的量而言,以作为其残炭物(非晶质碳)的重量比(〔[非晶质碳的重量]/[石墨的重量]〕
×
100)计,通常为0.01%以上、优选为0.1%以上、更优选为0.5%以上、进一步优选为1%以上、特别优选为2%以上、最优选为3%以上,另一方面,优选为30%以下、更优选为20%以下、进一步优选为15%以下、特别优选为10%以下。如果该重量比为上述范围,则为高容量、且li离子容易插入/脱离,因此低温输入输出特性、快速充放电特性及循环特性优异,从这点来看是优选的。
[0339]
<b3
‑
4.其它处理>
[0340]
为了制造本发明的一个实施方式的负极材料,可以对通过前述的制造方法得到的复合粒子另行进行粉碎处理。
[0341]
作为粉碎处理所使用的粗粉碎机,可列举颚式破碎机、冲击式压碎机、锥形压碎机等,作为中间粉碎机,可列举辊式压碎机、锤磨机等,作为微粉碎机,可列举球磨、振动磨、销棒粉碎机、搅拌磨、喷射磨等。这些当中,从粉碎时间短、处理速度的观点出发,优选球磨、振动磨。
[0342]
粉碎速度根据装置的种类、大小适宜设定,例如,球磨的情况下,通常为50rpm以上、优选为100rpm以上、更优选为150rpm以上、进一步优选为200rpm以上,另外,通常为2500rpm以下、优选为2300rpm以下、更优选为2000rpm以下。如果速度过快,则存在控制粒径变得困难的倾向,如果速度过慢,则存在处理速度变慢的倾向。
[0343]
粉碎时间通常为30秒钟以上、优选为1分钟以上、更优选为1分30秒钟以上、进一步优选为2分钟以上,另外,通常为3小时以下、优选为2.5小时以下、更优选为2小时以下。如果粉碎时间过短,则存在粒径控制变得困难的倾向,如果粉碎时间过长,则存在生产性下降的倾向。
[0344]
振动磨的情况下,粉碎速度通常为50rpm以上、优选为100rpm以上、更优选为150rpm以上、进一步优选为200rpm以上,另外,通常为2500rpm以下、优选为2300rpm以下、更优选为2000rpm以下。如果速度过快,则存在粒径的控制变得困难的倾向,如果速度过慢,则存在处理速度变慢的倾向。
[0345]
粉碎时间通常为30秒钟以上、优选为1分钟以上、更优选为1分30秒钟以上、进一步优选为2分钟以上,另外,通常为3小时以下、优选为2.5小时以下、更优选为2小时以下。如果粉碎时间过短,则存在粒径控制变得困难的倾向,如果粉碎时间过长,则存在生产性下降的倾向。
[0346]
为了制造本发明的一个实施方式的负极材料,可以对通过前述的制造方法得到的复合粒子进行粒径的分级处理。作为分级处理条件,可以使用开孔通常为53μm以下、优选为45μm以下、更优选为38μm以下的装置来实施。
[0347]
作为用于分级处理的装置,没有特别限制,例如,在干式筛分的情况下,可以使用旋转式筛、摇动式筛、转动式筛、振动式筛等,在干式气流式分级的情况下,可以使用重力式分级机、惯性力式分级机、离心力式分级机(分粒器、旋风分离器)等,在湿式筛分的情况下,可以使用机械式湿式分级机、水力分级机、沉降分级机、离心式湿式分级机等。
[0348]
〔b4.非水二次电池用负极〕
[0349]
本发明的一个实施方式的非水二次电池用负极(以下,有时称为“本发明的负极”)具备:集电体、和形成在该集电体上的活性物质层,其中,该活性物质层含有上述的本发明的一个实施方式的负极材料。本实施方式的非水二次电池用负极除了活性物质层以外,可以与第1实施方式同样。
[0350]
使用上述负极材料制作负极时,利用水性或有机类介质将在负极材料中配合有粘结树脂的材料制成浆料,并根据需要而向其中加入增稠剂后涂布在集电体、进行干燥即可。
[0351]
作为粘结树脂,优选使用相对于非水电解液稳定、且非水溶性的树脂。例如可使用:丁苯橡胶、异戊二烯橡胶及乙丙橡胶等橡胶状高分子;聚乙烯、聚丙烯、聚对苯二甲酸乙二醇酯、聚酰亚胺、聚丙烯酸、及芳香族聚酰胺等合成树脂;苯乙烯
‑
丁二烯
‑
苯乙烯嵌段共聚物或其加氢产物、苯乙烯
‑
乙烯
‑
丁二烯、苯乙烯共聚物、苯乙烯
‑
异戊二烯及苯乙烯嵌段共聚物及其氢化物等热塑性弹性体;间规立构
‑
1,2
‑
聚丁二烯、乙烯
‑
乙酸乙烯酯共聚物、及乙烯与碳原子数3~12的α
‑
烯烃的共聚物等软质树脂状高分子;四氟乙烯
‑
乙烯共聚物、聚偏氟乙烯、聚五氟丙烯及聚六氟丙烯等氟化高分子等。作为有机类介质,可使用例如n
‑
甲基吡咯烷酮及二甲基甲酰胺。
[0352]
相对于负极材料100重量份,通常使用0.1重量份以上、优选使用0.2重量份以上的粘结树脂。通过使粘结树脂的使用量相对于负极材料100重量份为0.1重量份以上,负极材料相互间的粘结力、负极材料与集电体的粘结力变得充分,可以防止由于从负极剥离负极材料而引起的电池容量的减少及循环特性的变差。
[0353]
另外,粘结树脂的使用量相对于负极材料100重量份优选为10重量份以下、更优选
为7重量份以下。通过使粘结树脂的使用量相对于负极材料100重量份为10重量份以下,可以防止负极容量的减少、且防止锂离子等碱金属离子相对于负极材料的出入受到阻碍等问题。
[0354]
作为添加到浆料中的增稠剂,可列举例如:羧甲基纤维素、甲基纤维素、羟乙基纤维素及羟丙基纤维素等水溶性纤维素类、聚乙烯醇及聚乙二醇等。这些当中,优选羧甲基纤维素。优选以相对于负极材料100重量份通常为0.1~10重量份、特别是0.2~7重量份来使用增稠剂。
[0355]
作为负极集电体,使用目前已知可用于该用途的例如铜、铜合金、不锈钢、镍、钛及碳等即可。集电体的形状通常为片状,还优选使用其表面带有凹凸的材料、网状物及冲孔金属等。
[0356]
在集电体上涂布负极材料和粘结树脂的浆料并进行干燥之后,优选进行加压而使形成在集电体上的活性物质层的密度增大、从而使负极活性物质层的每单位体积的电池容量增大。活性物质层的密度优选在1.2~1.8g/cm3的范围、更优选为1.3~1.6g/cm3。通过使活性物质层的密度为上述下限值以上,可以防止伴随着电极厚度的增大而带来的电池容量的降低。另外,通过使活性物质层的密度为上述上限值以下,伴随电极内的粒子间空隙减少而导致空隙中所保持的电解液量减少,可以防止锂离子等碱金属离子的移动性变小而使快速充放电特性降低。
[0357]
〔b5.非水二次电池〕
[0358]
本发明的一个实施方式的非水二次电池是具备正极、负极、以及电解质的非水二次电池,其中,作为负极,使用的是本发明的一个实施方式的负极。特别是,本发明的一个实施方式的非水二次电池中使用的正极及负极通常优选为能够吸留和放出li离子的正极及负极,本发明的一个实施方式的非水二次电池优选为锂离子二次电池。
[0359]
本发明的一个实施方式的非水二次电池除了使用了上述的本发明的一个实施方式的负极以外,可以按照常规方法来制造。特别是,本发明的一个实施方式的非水二次电池优选将[负极的容量]/[正极的容量]的值设计成1.01~1.5、更优选设计成1.2~1.4。
[0360]
[b5
‑
1.正极]
[0361]
作为本发明的一个实施方式的非水二次电池的成为正极的活性物质的正极材料,例如使用基本组成以licoo2表示的锂钴复合氧化物、以linio2表示的锂镍复合氧化物、以limno2及limn2o4表示的锂锰复合氧化物等锂过渡金属复合氧化物、二氧化锰等过渡金属氧化物、以及这些复合氧化物的混合物等即可。进一步,可使用tis2、fes2、nb3s4、mo3s4、cos2、v2o5、cro3、v3o3、feo2、geo2及lini
0.33
mn
0.33
co
0.33
o2、lifepo4等。
[0362]
可以通过将在上述正极材料中配合有粘结树脂的材料利用适当的溶剂进行浆料化并涂布在集电体上、并进行干燥而制作正极。需要说明的是,在浆料中,优选含有乙炔黑及科琴黑等导电材料。另外,也可以根据需要而含有增稠剂。需要说明的是,作为粘结材料及增稠剂,可以使用其用途公知的那些,例如可以使用作为用于负极的制造而示例出的那些。
[0363]
相对于正极材料100重量份,导电材料的配合量优选为0.5重量份~20重量份、更优选为1重量份~15重量份。另外,相对于正极材料100重量份,增稠剂的配合量优选为0.2重量份~10重量份、更优选为0.5重量份~7重量份。此外,就相对于正极材料100重量份的
粘结树脂的配合量而言,在用水将粘结树脂浆料化的情况下,优选为0.2重量份~10重量份、更优选为0.5重量份~7重量份,另一方面,在用n
‑
甲基吡咯烷酮等溶解粘结树脂的有机溶剂将粘结树脂浆料化的情况下,优选为0.5重量份~20重量份、更优选为1~15重量份。
[0364]
作为正极集电体,可列举例如:铝、钛、锆、铪、铌及钽等、以及它们的合金。这些当中,优选铝、钛及钽、及其合金,最优选为铝及其合金。
[0365]
[b6
‑
2.电解液]
[0366]
电解液可使用在以往周知的非水溶剂中溶解各种锂盐而成的溶液。
[0367]
作为非水溶剂,可使用例如碳酸亚乙酯、氟代碳酸亚乙酯、碳酸亚丙酯、碳酸亚丁酯及碳酸亚乙烯酯等环状碳酸酯;碳酸二甲酯、碳酸甲乙酯及碳酸二乙酯等链状碳酸酯;γ
‑
丁内酯等环状酯;冠醚、2
‑
甲基四氢呋喃、四氢呋喃、1,2
‑
二甲基四氢呋喃及1,3
‑
二氧戊环等环状醚;1,2
‑
二甲氧基乙烷等链状醚等。通常可以将这些溶剂中的2种以上混合使用。这些当中,优选使用环状碳酸酯和链状碳酸酯,或者在其中进一步混合其它溶剂后使用。
[0368]
也可以在电解液中添加碳酸亚乙烯酯、乙烯基碳酸亚乙酯、琥珀酸酐、马来酸酐、丙磺酸内酯及二乙基砜等化合物、二氟磷酸锂这样的二氟磷酸盐等。进一步,还可以添加二苯基醚及环己基苯等过充电防止剂。
[0369]
作为溶解于非水溶剂的电解质,可列举例如:liclo4、lipf6、libf4、licf3so3、lin(cf3so2)2、lin(cf3cf2so2)2、lin(cf3so2)(c4f9so2)及lic(cf3so2)3等。电解液中的电解质的浓度通常为0.5mol/l~2mol/l、优选为0.6mol/l~1.5mol/l。
[0370]
[b6
‑
3.隔板]
[0371]
本发明的一个实施方式的非水二次电池中优选使用隔板,且该隔板介于正极和负极之间。作为这样的隔板,优选使用聚乙烯、聚丙烯等聚烯烃的多孔性片、或无纺布。
[0372]
<c.第3实施方式>
[0373]
以下,对本发明进行详细说明,但本发明并不限定于以下说明,可以在不脱离本发明主旨的范围内任意变形后实施。需要说明的是,在本发明中,使用“~”并在其前后夹着数值或物性值地表现的情况下,使用的是包含其前后的值的含义。
[0374]
〔c1.非水二次电池用负极材料〕
[0375]
本发明的一个实施方式的非水二次电池用负极材料(以下,有时简称为“负极材料”)是以下的非水二次电池用负极材料:其包含在表面的至少一部分具有非晶质碳物质或石墨质物质中的至少一者的石墨,该非水二次电池用负极材料通过压汞法测定的微孔分布具有2个以上的峰,将微孔径为最小的峰与下一个峰的谷间的极小值以下的累积孔容设为y[ml/g]、将石墨表面的非晶质碳物质及石墨质物质的覆盖率设为x(%)时,满足下述式(1)及(2),所述微孔径为最小的峰的峰顶的微孔径为360nm以下。
[0376]
式(1):y>0.005
[0377]
式(2):y<
‑
0.006x+0.12
[0378]
另外,本发明的另一实施方式的非水二次电池用负极材料优选包含在表面的至少一部分具有非晶质碳物质或石墨质物质中的至少一者的石墨,该非水二次电池用负极材料通过压汞法测定的微孔分布具有2个以上的峰,将其峰的谷间的极小值以下的累积孔容设为y[ml/g]、将覆盖率设为x(%)时,满足下述式(1)及(3)。
[0379]
式(1):y>0.005
[0380]
式(3):y<
‑
0.006x+0.079
[0381]
本实施方式的非水二次电池用负极材料发挥可提供面向高容量的电池的能够以小的压制负载而压制加工至高密度、且极板膨胀小的非水二次电池。本发明发挥这样的效果的理由虽然尚不确定,但认为是由于以下理由。
[0382]
在本实施方式的负极材料中,利用压汞法求出的孔容中的在极小值以下的微孔径的范围中的累积孔容成为存在于负极材料粒子内部的孔容的指标。通过将其减小至相对于覆盖率为特定的范围,在将电极进行压制而达到一定密度时,与以往的存在于粒子内部的孔容大的负极材料相比,能够以粒子的变形更小的状态进行压制,与完全没有粒子内微孔的负极材料相比,进行压制时能够适度地变形。其结果,能够压制成更高密度。
[0383]
此外,压汞法中最小的峰成为粒子内微孔的尺寸的指标。该粒子内的微孔的峰小表示的是,微孔径小的微孔存在于粒子内,粒子内的鳞片彼此间更致密地粘结在一起。认为通过致密地粘结,循环后的极板膨胀减小。
[0384]
[c1
‑
1.物性]
[0385]
本发明的一个实施方式的负极材料满足以下说明的累积孔容与覆盖率的关系。另外,本发明的负极材料优选满足以下的各物性。
[0386]
<c1
‑1‑
1.孔容>
[0387]
本发明的负极材料中,通过压汞法(汞孔隙率法)测定的微孔分布具有2个以上的峰,且将微孔径为最小的峰与下一个峰的谷间的极小值以下的累积孔容设为y(ml/g)、将覆盖率设为x(%)时,满足下述式(1)及(2)。
[0388]
式(1):y>0.005
[0389]
式(2):y<
‑
0.006x+0.12
[0390]
峰的数量为2个以上、更优选为2个。
[0391]
微孔分布的峰表示的是粒子间的空隙和粒子内的空隙,峰为一个的情况下,意味着仅存在粒子间空隙,进行压制时的变形变得不充分。
[0392]
作为式(1),由于需要存在粒子内空隙,因此为y>0.005、更优选为y>0.01、进一步优选为y>0.015。
[0393]
作为式(2),在设为y<αx+β的情况下,将横轴设为覆盖率x、将上述累积孔容y设为y轴,表示图1中的式(2)的斜率,β表示式(2)的y截距。从非晶质碳物质及石墨质物质的覆盖率和填埋粒子内空隙的效率方面出发,α优选为
‑
0.0055以下,且优选为
‑
0.007以上、更优选为
‑
0.0065以上、最优选为
‑
0.006以上。从用非晶质碳物质或石墨质物质中的至少一者覆盖之前的粒子内空隙的值方面出发,β优选为0.11以下、更优选为0.10以下、进一步优选为0.09以下、特别优选为0.079以下、最优选为0.07以下。为上述式的范围外表示的是,微孔未被非晶质碳物质或石墨质物质中的至少一者良好地覆盖,从而无法得到良好的压制性,作为结果,对快速充电、输入输出保存特性带来不良影响。
[0394]
微孔径小的一侧的峰顶表示的是粒子内微孔的尺寸小。粒子内微孔细表示的是例子内存在微孔径小的微孔、粒子内的鳞片彼此间进一步致密化,从这方面出发,用压汞法测定的孔容中微孔径为最小的峰(从小的一侧的峰)中的峰顶的微孔径优选为350nm以下、更优选为280nm以下、进一步优选为220nm以下、更进一步优选为160nm以下、特别优选为100nm以下、最优选为85nm以下。需要说明的是,峰仅为1个的情况下,表示的是不存在粒子内空
隙。小的一侧的峰处于上述范围外表示粒子内以较高的频度存在大的空隙,显示出形成粒子的鳞片彼此间的粘结力较小。其结果,在反复进行充放电时,粒子发生膨胀,结果极板可能进一步膨胀。
[0395]
<c1
‑1‑
2.累积孔容>
[0396]
在本发明的负极材料中,微孔分布的微孔径小的一侧的2个峰的谷间的极小值以下的范围中的累积孔容是使用压汞法(汞孔隙率法)测得的值,其通常为0.12ml/g以下、优选为0.080ml/g以下、更优选为0.070ml/g以下、进一步优选为0.060ml/g以下、特别优选为0.050ml/g以下、最优选为0.040ml/g以下,另一方面,优选为0.001ml/g以上、更优选为0.002ml/g以上、进一步优选为0.005ml/g以上、特别优选为0.010ml/g以上、最优选为0.020ml/g以上。如果为上述范围内,则充放电使li离子能够在电极内顺利地移动,快速充放电特性、低温输入输出特性良好,因此优选。
[0397]
作为用于上述汞孔隙率法的装置,可以使用水银孔隙率仪(autopore9520:micrometritics公司制造)。称量试样(负极材料)使其为0.2g左右的值,封入到粉末用容器中,在室温、真空下(50μmhg以下)进行10分钟脱气来实施前处理。接着,减压至4psia(约28kpa),向上述容器中导入水银,使压力从4psia(约28kpa)阶梯状地升压至40000psia(约280mpa),然后降压至25psia(约170kpa)。升压时的阶梯数设为80点以上,在各阶梯中保持10秒钟的平衡时间后,测定水银压入量。使用washburn式由这样得到的水银压入曲线计算出微孔分布。需要说明的是,以水银的表面张力(γ)为485dyne/cm、接触角(ψ)为140
°
来进行计算。
[0398]
然后,基于所得到的结果,以横轴为微孔径、纵轴为孔容进行作图,求出该图表中微孔径最小的峰与下一个峰的谷间的极小值,将该极小值以下定义为累积孔容y。
[0399]
<c1
‑1‑
3.覆盖率>
[0400]
在本说明书中,覆盖率x(%)可以按照以下方式求出。即,可以基于石墨与非晶质前体的混合比率、以及烧成后的烧成产率来进行计算。
[0401]
式(4):
[0402]
覆盖率x(%)=([烧成后的样品重量-石墨的重量]/[烧成后的样品重量]
×
100)
[0403]
另外,混合比率、烧成产率不明的样品的情况下,可以利用非晶质与石墨的真密度之差来进行推测。
[0404]
首先,根据xrd的d002值来确认母材石墨的结晶性。石墨的理论的d002值为结晶性高的天然石墨显示出接近于理论值的值。另一方面,对于人造石墨而言,根据原料焦炭的种类、石墨化温度的不同,其d002的数值有较大变动。d002值优选为以下、更优选为以下、进一步优选为d002值为上述范围外时,石墨的结晶性低、不具有足够的充放电容量,因此不优选。如果能够确认是xrd的d002值为以下的高结晶,则能够使用下述推定式(5)来推测覆盖率。
[0405]
式(5):
[0406]
覆盖率(%)=596.72-264.02
×
真密度
[0407]
本发明的一个实施方式的负极材料的真密度优选为2.200g/cm3以上、更优选为2.210g/cm3以上、进一步优选为2.200g/cm3以上,石墨的理论真密度为2.262g/cm3。
[0408]
上述覆盖率通常为20%以下、优选为15%以下、更优选为10%以下、进一步优选为9%以下、特别优选为8%以下、最优选为7%以下,另一方面,优选为0.1%以上、更优选为1%以上、进一步优选为2%以上、特别优选为3%以上。如果为上述上限以下,则负极材料中石墨的比例适量,容易进行高容量化,另一方面,如果为下限以上,则非晶质适量,可以将锂离子从石墨表面顺利地移动,因此快速充放电特性、低温输入输出特性良好。
[0409]
真密度是以通过使用了丁醇的液体置换法(比重瓶法)测定的值定义的。为了尽可能降低测定时的测定误差,优选使测定次数为至少3次、优选为5次、进一步优选为7次、特别优选为10次来实施并使用其平均值。
[0410]
本发明的碳材料的d002值可以通过基于学振法的x射线衍射求出。这里的x射线衍射测定条件如下所述。
[0411]
x射线:cu kα射线
[0412]
测定范围、及步距角:20度≤2θ≤30度、0.013度
[0413]
试料调整:在深度0.2mm的试料板凹部填充粉末试料而制作平坦的试料面,作为通过上述x射线衍射测定的面间距d002及微晶尺寸lc002,可以使用按照学振法测定的值。需要说明的是,在学振法中,对于100nm以上的值不进行区别,均记作
[0414]
<c1
‑1‑
4.bet比表面积(sa)>
[0415]
本发明的一个实施方式的负极材料的基于bet法的比表面积(sa)优选为0.5m2/g以上、更优选为0.8m2/g以上、进一步优选为1.0m2/g以上,另一方面,优选为10.0m2/g以下、更优选为6.5m2/g以下、进一步优选为5.0m2/g以下、特别优选为4.0m2/g以下。如果sa为上述下限值以上,则可确保li离子进出的部位,存在锂离子二次电池的快速充放电特性、低温输入输出特性变得良好的倾向。另一方面,如果sa为上述上限值以下,则活性物质对于电解液的活性变得过于过剩,与电解液的副反应被抑制而防止电池的初始充放电效率的降低、气体产生量的增加,存在电池容量提高的倾向。
[0416]
需要说明的是,bet比表面积(sa)可以使用mountech公司制造的macsorb来进行测定。具体可以如下测定:对于试料在氮流通下于100℃进行了3小时预减压干燥后,使用冷却至液氮温度、且氮气相对于大气压的相对压力值准确调节为0.3的氮氦混合气体,通过采用气体流动法的氮吸附bet1点法来进行测定。
[0417]
<c1
‑1‑
5.振实密度>
[0418]
本发明的一个实施方式的负极材料的振实密度(g/cm3)优选为1.15g/cm3以上、更优选为1.16g/cm3以上、进一步优选为1.17g/cm3以上、特别优选为1.18g/cm3以上、最优选为1.20g以上,另一方面,优选为1.40g/cm3以下、更优选为1.35g/cm3以下、进一步优选为1.30g/cm3以下。如果振实密度为上述下限值以上,则在制作极板时的条痕产生等的工序性变得良好,负极材料层的填充性提高,因此,压延性良好、容易形成高密度的负极片,高密度化成为可能,制成电极体时li离子迁移路径的弯曲度变小、且粒子间空隙的形状整齐,因此电解液的移动变得顺利,快速充放电特性提高,从上述观点出发是优选的,另外,如果为上述上限值以下,则粒子的表面及内部具有适度的空间,粒子不会变得过硬,电极压制性优异,并且低温输入输出特性、快速充放电特性优异,从上述观点出发是优选的。
[0419]
需要说明的是,振实密度使用粉体密度测定器tapdenser kyt
‑
3000(seishin enterprise株式会社制)进行测定。具体地,使试料落入20cc的振实容器中,在填充满容器之后,进行1000次冲程长度为10mm的振实,将此时的密度作为振实密度。
[0420]
<c1
‑1‑
6.压片密度@2.4t/cm3>
[0421]
在内径φ10的模具中插入作为挤压工具的φ10、长度35mm的轴、以及作为支撑工具的φ10、长度6mm的轴这2种工具,然后安装于能够测定夹入时的载荷和高度(厚度)的装置(例如,mitsubishi chemical analytech co ltd.制粉体抵抗测定系统),用油压泵施加15kgf的载荷,测定工具高度。接下来,仅取出挤压工具并加入0.6g复合粒子,再次插入挤压工具。将模具安装于油压千斤顶(例如,as one公司制高压千斤顶j
‑
1),关紧压力阀缓慢加压至0.9t/cm2,再迅速加压至2.4t/cm2后,保持3秒钟,将手从油压千斤顶离开,等待60秒钟,松开压力阀进行减压。然后,再次安装于能够测定夹入时的载荷和高度的装置,用油压泵施加15kgf的载荷,测定加压后的工具高度。同时测定加压后的复合粒子的重量,将由前述的工具高度的差值和重量计算出的密度定义为压片密度。没单位面积的载荷可以由油压千斤顶的刻度和圆柱体直径、模具的内径算出。例如,在后述的实施例中,使用φ22mm的圆柱体直径,进行加压直至轴的刻度达到500kgf,施加了2.4t/cm2的载荷。
[0422]
<c1
‑1‑
7.压片密度-振实密度>
[0423]
由压片密度减去振实密度(g/cm3)而得到的数值表示的是施加载荷时的压密容易程度,可以作为粒子的硬度的指标而使用。
[0424]
压片密度-振实密度优选为0.1g/cm3以上、更优选为0.15g/cm3以上、进一步优选为0.2g/cm3以上、特别优选为0.26g/cm3以上,另一方面,优选为0.8g/cm3以下、更优选为0.6g/cm3以下、进一步优选为0.4g/cm3以下。如果压片密度-振实密度为上述上限值以下,则粒子具有适度的硬度,在压制成高密度时电极表面的粒子不会过于破坏,电解液的移动变得顺畅,因此优选。另外,如果为上述下限以上,则粒子不会过硬,能够压制成高密度,因此优选。
[0425]
<c1
‑1‑
8.体积基准平均粒径(平均粒径d50)>
[0426]
本发明的一个实施方式的负极材料的体积基准平均粒径(也记作“平均粒径d50”)优选为1μm以上、更优选为3μm以上、进一步优选为4μm以上、特别优选为5μm以上,另外,优选为50μm以下、更优选为40μm以下、进一步优选为30μm以下、特别优选为25μm以下、最优选为20μm以下。如果d50的值为上述下限值以上,则存在可以防止不可逆容量的增加、初始电池容量的损失的倾向,另一方面,如果d50的值为上述上限值以下,则可防止浆料涂布中的条痕等工序不良情况的产生,存在快速充放电特性、低温输入输出特性提高的倾向。
[0427]
平均粒径(d50)如下定义:使0.01g复合粒子悬浮于作为表面活性剂的聚氧乙烯山梨糖醇酐单月桂酸酯(作为例子,可以举出tween 20(注册商标))的0.2质量%水溶液10ml中,将其作为测定样品导入到市售的激光衍射/散射式粒度分布测定装置(例如horiba制造的la
‑
920)中,对测定样品以60w的输出功率照射28khz的超声波1分钟后,在上述测定装置中测定体积基准的中位径,将其定义为平均粒径d50。
[0428]
[c2.制造方法]
[0429]
本发明的一个实施方式的非水二次电池用负极材料的制造方法只要是能够制造包含在表面的至少一部分具有非晶质碳物质或石墨质物质中的至少一者、满足相对于覆盖
率的特定的粒子内空隙的关系、且通过压汞法测得的孔容中极小值以下的峰的峰顶满足特定的大小的负极材料的方法,就没有特别限制,例如,可以通过对球状石墨进行加压处理,并与非晶质碳前体(非晶质碳物质的原料)混合、烧成来进行制造,所述球状石墨是将鳞片细细地粉碎后,在造粒材料的存在下实施球形化处理而得到的。加压处理可列举各向异性、各向同性的加压处理,但从将上述累积孔容y与上述覆盖率x的关系控制在特定的范围内的观点出发,优选各向同性加压处理。另外,作为加压处理的条件,没有特别限定,通过在50mpa以上且300mpa以下进行处理,可以将上述累积孔容y与上述覆盖率x的关系控制在特定的范围内。对于加压处理的条件而言,从将上述累积孔容y与上述覆盖率x的关系控制在特定的范围内的观点出发,优选为100mpa以上、更优选为120mpa以上、进一步优选为140mpa以上、特别优选为160mpa以上、最优选为180mpa以上,另外,优选为280mpa以下、更优选为260mpa以下、进一步优选为240mpa以下、特别优选为230mpa以下、最优选为220mpa以下。上述的制造方法具体来说由于能够通过控制鳞片的粉碎条件、球形化条件、成型压力、覆盖率来控制每单位覆盖率的孔容、并且能够控制小的峰的峰顶的大小,因此优选。
[0430]
<c2
‑
1.石墨>
[0431]
本发明的一个实施方式的负极材料包含石墨。用于制造本发明的一个实施方式的负极材料的石墨优选显示以下的种类、物性者。需要说明的是,关于石墨的物性,只要未对其测定条件及定义进行特别说明,则与前述的对于负极材料进行了说明的那些同样。
[0432]
对于石墨的种类而言,只要是能够吸留和放出锂离子即可,其种类没有特别限定,可以是天然石墨、人造石墨中的任意石墨。作为天然石墨,可以是鳞片状石墨、块状石墨、土壤石墨等的任意石墨,但优选杂质少的石墨,优选根据需要实施公知的纯化处理后使用。作为人造石墨,可列举:对煤焦油沥青、煤炭类重油、常压渣油、石油类重油、芳香烃、含氮环状化合物、含硫环状化合物、聚苯、聚氯乙烯、聚乙烯醇、聚丙烯腈、聚乙烯醇缩丁醛、天然高分子、聚苯硫醚、聚苯醚、糠醇树脂、苯酚酚醛树脂、酰亚胺树脂等有机物在通常为2500℃以上、且通常为3200℃以下的范围的温度进行烧成、石墨化而得到的人造石墨。此时,也可以将含硅化合物、含硼化合物等用作石墨化催化剂。
[0433]
对于石墨的结晶性(石墨化度)而言,通过基于x射线广角衍射法的学振法求出的(002)面的面间距(d002)通常优选为以下、更优选为以下、进一步优选为以下。d002为上述范围内时,石墨的结晶性适宜,具有足够的充放电容量,是优选的。
[0434]
对于石墨的形状而言,从快速充放电特性的观点出发,特别优选为球状石墨(球状化石墨)。作为对石墨粒子进行球状化的方法,具体优选具有造粒工序,该造粒工序是至少赋予冲击、压缩、摩擦及剪切力中的任意的力学能来对造粒原料碳材料的工序,且上述造粒工序在满足下述1)及2)的条件的造粒剂存在下进行。
[0435]
1)在造粒前述原料碳材料的工序中,造粒材料为液体。
[0436]
2)造粒材料包含成为非晶质碳的有机化合物。
[0437]
3)造粒剂不含有机溶剂,或者在包含有机溶剂的情况下,有机溶剂中的至少一种不具有闪点、或者在具有闪点时该闪点为5℃以上。
[0438]
只要具有上述造粒工序即可,也可以根据需要进一步具有其它工序。其它工序可以单独实施,也可以同时实施多个工序。作为一个实施方式,可列举包含以下的第1工序至
第5工序的方式。以下,对这些工序进行说明。
[0439]
(第1工序)调整原料碳材料的粒度的工序
[0440]
(第2工序)将原料碳材料和造粒剂混合的工序
[0441]
(第3工序)对原料碳材料进行造粒的工序
[0442]
(第4工序)除去造粒剂的工序
[0443]
(第5工序)在造粒碳材料中添加附着结晶性低于原料碳材料的非晶质碳物质的工序
[0444]
(第1工序)调整原料碳材料的粒度的工序
[0445]
用于本发明的非水二次电池用负极材料的制造的原料碳材料使用前述的石墨。
[0446]
原料碳材料优选在第1工序中调整为下面这样的粒度。即,得到的原料碳材料的平均粒径(d50)优选为1μm以上、更优选为2μm以上、进一步优选为3μm以上,另一方面,优选为30μm以下、更优选为20μm以下、进一步优选为15μm以下、特别优选为12μm以下、最优选为9μm以下。
[0447]
通过调整原料碳材料的粒度使得d50为上述范围内,在球形化处理后成为作为负极材料而言理想的粒径,并且鳞片彼此间的距离变短,成为强固的粒子,因此优选。
[0448]
作为将原料碳材料的d50调整为上述范围的方法,可列举例如对(天然)石墨粒子进行粉碎或分级中的至少一者的方法。
[0449]
粉碎所使用的装置没有特别限制,例如,作为粗粉碎机,可列举剪切式磨、颚式破碎机、冲击式压碎机、锥形压碎机等,作为中间粉碎机,可列举辊式压碎机、锤磨机等,作为微粉碎机,可列举机械式粉碎机、气流式粉碎机、旋流式粉碎机等。具体可列举球磨、振动磨、销棒粉碎机、搅拌磨、喷射磨、旋风磨、涡轮磨等。特别是,在欲得到d50为10μm以下的石墨粒子的情况下,优选使用气流式粉碎机、旋流式粉碎机。
[0450]
作为用于分级处理的装置,没有特别限制,例如,在干式筛分的情况下,可以使用旋转式筛、摇动式筛、转动式筛、振动式筛等,在干式气流式分级的情况下,可以使用重力式分级机、惯性力式分级机、离心力式分级机(分粒器、旋风分离器)等,另外,进行湿式筛分的情况下,可以使用机械式湿式分级机、水力分级机、沉降分级机、离心式湿式分级机等。
[0451]
另外,作为第一工序中得到的原料碳材料,优选满足以下的物性。
[0452]
对于原料碳材料的通过x射线广角衍射法测得的002面的面间距(d002)及微晶尺寸(lc)而言,通常d002为以下、lc为以上,优选d002为以下、lc为以上。d002及lc是表示碳材料主体(bulk)的结晶性的值,d002的值越小、并且lc越大,表示为结晶性高的碳材料,由于进入石墨层间的锂量接近于理论值,因此容量增加。如果结晶性低,则存在无法表现出将高结晶性石墨用于电极的情况下的高容量、且不可逆容量低这样的优异的电池特性的倾向。d002和lc特别优选上述范围的组合。
[0453]
x射线衍射通过以下方法来测定。首先,在碳粉末中加入总量的约15质量%的x射线标准高纯度硅粉末并进行混合,将混合后的混合物作为材料,以用石墨单色器进行了单色化的cukα射线作为射线源,用反射式x射线衍射仪法测定广角x射线衍射曲线。然后,使用学振法求出面间距(d002)及微晶尺寸(lc)。
[0454]
原料碳材料的填充结构受到粒子的大小、形状、粒子间相互作用力的程度等的左右,但在本发明中,还可以应用振实密度作为定量地讨论填充结构的指标之一。经本发明人
等的研究确认,对于真密度和平均粒径基本相等的石墨质粒子而言,越是形状为球状、粒子表面越平滑,振实密度越是显示高值。即,为了提高振实密度,重要的是使粒子的形状接近于球状、并保持粒子表面的平滑程度。粒子形状接近球状、粒子表面平滑时,粉体的填充性也显著提高。原料碳材料的振实密度优选为0.1g/cm3以上、更优选为0.15g/cm3以上、进一步优选为0.2g/cm3以上、特别优选为0.3g/cm3以上。振实密度通过实施例中的后面所述的方法来测定。
[0455]
原料碳材料的氩离子激光拉曼光谱被用作表征粒子表面性状的指标。原料碳材料的拉曼r值优选为0.05以上且0.9以下,更优选为0.05以上且0.7以下,进一步优选为0.05以上且0.5以下,所述拉曼r值是氩离子激光拉曼光谱中1360cm
‑1附近的峰强度相对于1580cm
‑1附近的峰强度的比值。r值是表示碳粒子表面附近(距粒子表面以内)的结晶性的指标,r值越小,表示结晶性越高,或者结晶状态未发生紊乱。拉曼光谱通过以下所示的方法来测定。具体地,通过使测定对象粒子自然落下到拉曼分光器测定池内来进行试料填充,对测定池内照射氩离子激光,并使测定池在与该激光垂直的面内旋转来进行测定。需要说明的是,氩离子激光的波长为514.5nm。
[0456]
原料的鳞片状石墨的bet比表面积(sa)优选为5m2/g以上、更优选为10m2/g以上,且优选为25m2/g以下、更优选为20m2/g以下。如果为上述范围内,则在下一工序的球形化工序中可有效地被球形化,因此优选。
[0457]
原料的鳞片状石墨的bet比表面积(sa)可以使用mountech公司制造的macsorb来进行测定。具体可以如下测定:对于试料在氮流通下于100℃进行了3小时预减压干燥后,使用冷却至液氮温度、且氮气相对于大气压的相对压力值准确调节为0.3的氮氦混合气体,通过采用气体流动法的氮吸附bet 1点法来进行测定。
[0458]
(第2工序)将原料碳材料和造粒剂混合的工序
[0459]
本发明的一个实施方式中使用的造粒剂满足下述条件:1)在上述对原料碳材料进行造粒的工序时为液体;2)造粒材料包含成为非晶质碳的有机化合物;以及3)造粒剂不含有机溶剂,或者在包含有机溶剂的情况下,有机溶剂中的至少一种不具有闪点、或者在具有闪点时该闪点为5℃以上。
[0460]
通过具有满足上述要件的造粒剂,在接下来的第3工序的对原料碳材料进行造粒的工序时,通过造粒剂在原料碳材料间进行液体架桥,在原料碳材料间液桥内的毛细管负压和液体表面张力作用下而产生的引力在粒子间发挥液体架桥附着力的作用,原料碳材料间的液体架桥附着力增大,从而使原料碳材料更牢固地附着于粒子表面,能够更有效地缩短鳞片彼此间的距离,因此优选。
[0461]
在本发明中,造粒剂在原料碳材料间进行液体架桥产生的原料碳材料间的液体架桥附着力的强度与γcosθ值成比例(其中,γ:液体的表面张力、θ:液体与粒子的接触角)。即,在对原料碳材料进行造粒时,造粒剂优选与原料碳材料的润湿性高者,具体来说,优选选择cosθ>0的造粒剂,使得γcosθ值>0,造粒剂的通过下述测定方法测定的与石墨的接触角θ优选小于90
°
。
[0462]
(与石墨的接触角θ的测定方法)
[0463]
在hopg表面滴加1.2μl的造粒剂,用接触角测定装置(例如,协和界面株式会社制自动接触角测量仪dm
‑
501)测定润湿扩展收敛而使一秒钟的接触角θ的变化率达到3%以下
时(也称为稳定状态)的接触角。其中,在使用25℃下的粘度为500cp以下的造粒剂的情况下,将25℃下的值作为接触角θ的测定值,在使用25℃下的粘度大于500cp的造粒剂的情况下,将加温至粘度达到500cp以下的温度下的值作为接触角θ的测定值。
[0464]
此外,原料碳材料与造粒剂的接触角θ越接近于0
°
,γcosθ值越是变大,因此,石墨粒子间的液体架桥附着力增大,石墨粒子彼此能够更强固地附着。因此,上述造粒剂与石墨的接触角θ更优选为85
°
以下,进一步优选为80
°
以下,更进一步优选为50
°
以下,特别优选为30
°
以下,最优选为20
°
以下。
[0465]
通过使用表面张力(γ)大的造粒剂,γcosθ值也变大,石墨粒子的附着力提高,因此,γ优选为0以上,更优选为15以上,进一步优选为30以上。造粒剂的表面张力(γ)可以使用表面张力仪(例如,协和界面科学株式会社制dca
‑
700)、按照wilhelmy法来测定。
[0466]
另外,作为对于伴随着粒子的移动而产生的液桥的伸长的阻抗成分,粘性力发挥作用,其大小与粘度成比例。因此,如果在对原料碳材料进行造粒的造粒工序时为液体,则造粒剂的粘度没有特别限定,但优选在造粒工序时为1cp以上。
[0467]
造粒剂在25℃下的粘度优选为1cp以上且100000cp以下、更优选为5cp以上且10000cp以下、进一步优选为10cp以上且8000cp以下、特别优选为50cp以上且6000cp以下。如果粘度在上述范围内,则在对原料碳材料进行造粒时,可以防止由于与转子、壳体的碰撞等的冲击力而引起附着粒子的脱离。
[0468]
本发明中使用的造粒剂的粘度使用流变仪(例如rheometric scientific公司制ares)、将适量测定对象(这里为造粒剂)加入到杯中,调节为给定的温度后测定。在剪切速度100s
‑1下的剪切应力为0.1pa以上的情况下,将剪切速度100s
‑1下测定的值定义为本说明书中的粘度,在剪切速度100s
‑1下的剪切应力小于0.1pa的情况下,将1000s
‑1下测定的值定义为本说明书中的粘度,在剪切速度1000s
‑1下的剪切应力小于0.1pa的情况下,将在剪切应力达到0.1pa以上的剪切速度下测定的值定义为本说明书中的粘度。需要说明的是,也可以通过将所使用的轴设为适于低粘度流体的形状来使剪切应力为0.1pa以上。
[0469]
另外,造粒剂在上述将原料碳材料和造粒剂混合时的粘度优选为1cp以上且1000cp以下、更优选为5cp以上且800cp以下、进一步优选为10cp以上且600cp以下、特别优选为20cp以上且500cp以下。如果粘度在上述范围内,则造粒材料均匀地附着于原料碳材料,在对原料碳材料进行造粒时,能够防止由于与转子、壳体的碰撞等的冲击力而引起的附着粒子的脱离,另外,造粒材料进入到1nm至4nm的微孔中,在后工序中成为非晶质碳,由此,可以减少该微孔,从而能够制造低温输入输出特性、高温保存特性优异的负极材料,因此优选。上述的在对原料碳材料进行造粒的工序中的粘度可以通过后述的有机溶剂的添加、混合温度的控制来进行调整。
[0470]
另外,本发明的实施方式中使用的造粒剂包含成为非晶质碳的有机化合物。由此,通过造粒材料进入到1nm至4nm的微孔中而形成非晶质碳,能够减少该微孔,从而能够制造低温输入输出特性、高温保存特性优异的负极材料。例如可列举石油系、煤系的重油、焦油及沥青、聚乙烯醇、聚丙烯腈、酚醛树脂、纤维素等的树脂,根据需要,可以使用水系溶剂、或者使用不具有闪点的有机溶剂、或在具有闪点时使用闪点为5℃以上的有机溶剂等来进行稀释。这些当中,优选包含作为软碳的石油系、煤系的重油、焦油、沥青等,这是因为,它们作为非晶质碳时不易生成微孔、能够更有效地减少1nm至4nm的微孔。
[0471]
此外,本发明的实施方式中使用的造粒剂不含有机溶剂,或者在含有有机溶剂的情况下,有机溶剂中的至少一种不具有闪点,或者在具有闪点时该闪点为5℃以上。由此,在接下来的第3工序的对原料碳材料进行造粒时,可以防止冲击、放热所引发的造粒剂的起火、火灾及爆炸的危险,因此,可以稳定且高效地进行制造。
[0472]
作为闪点5℃以上的有机溶剂,可列举:液体石蜡等石蜡系油、烯烃系油、环烷烃系油、芳香族系油等合成油、植物系油脂类、动物系脂肪族类、酯类、高级醇类等天然油;二甲苯、异丙苯、乙苯、丙苯等烷基苯;甲基萘、乙基萘、丙基萘等烷基萘;苯乙烯等烯丙基苯、烯丙基萘等芳香烃类;辛烷、壬烷、癸烷等脂肪烃类;甲基异丁基酮、二异丁基酮、环己酮等酮类;乙酸丙酯、乙酸丁酯、乙酸异丁酯、乙酸戊酯等酯类;甲醇、乙醇、丙醇、丁醇、异丙醇、异丁醇、乙二醇、丙二醇、二乙二醇、三乙二醇、四乙二醇、甘油等醇类;乙二醇单甲醚、乙二醇单乙醚、乙二醇单丁醚、二乙二醇单丁醚、三乙二醇单丁醚、四乙二醇单丁醚、甲氧基丙醇、甲氧基丙基
‑2‑
乙酸酯、甲氧基甲基丁醇、甲氧基丁基乙酸酯、二乙二醇二甲基醚、二丙二醇二甲基醚、二乙二醇乙基甲基醚、三乙二醇二甲基醚、三丙二醇二甲基醚、四乙二醇二甲基醚、乙二醇单苯基醚等二醇类衍生物类;1,4
‑
二氧杂环己烷等醚类;二甲基甲酰胺、吡啶、2
‑
吡咯烷酮、n
‑
甲基
‑2‑
吡咯烷酮等含氮化合物;二甲亚砜等含硫有机化合物;二氯甲烷、氯仿、四氯化碳、二氯乙烷、三氯乙烷、氯苯等含卤有机化合物;以及它们的混合物等,不包含例如甲苯这样的闪点低的物质。这些有机溶剂也可以单独作为造粒剂使用。需要说明的是,在本说明书中,闪点可以通过公知的方法来测定。
[0473]
作为将原料碳材料和造粒剂进行混合的方法,可列举例如:使用混合器、捏合机将原料碳材料和造粒剂进行混合的方法、将使有机化合物溶解于低粘度稀释溶剂(有机溶剂)而得到的造粒剂与原料碳材料进行混合,然后再除去该稀释溶剂(有机溶剂)的方法等。这些方法中,优选使用混合器、捏合机将原料碳材料和造粒剂进行混合的方法,这是由于,该方法中,有机化合物中的成为碳物质的成分越多,越是能够更有效地减少1nm至4nm的微孔。另外,还可以举出在接下来的第3工序中对原料碳材料进行造粒时,同时进行将造粒剂和原料碳材料投入到造粒装置中来将原料碳材料和造粒剂进行混合的工序、以及进行造粒的工序的方法。
[0474]
相对于原料碳材料100重量份,造粒剂的添加量优选为0.1重量份以上、更优选为1重量份以上、进一步优选为3重量份以上、更进一步优选为6重量份以上、再进一步优选为10重量份以上、特别优选为12重量份以上、最优选为15重量份以上,且优选为1000重量份以下、更优选为100重量份以下、进一步优选为80重量份以下、特别优选为50重量份以下、最优选为30重量份以下。如果在上述范围内,则不易产生粒子间附着力降低而引起的球形化度的降低、原料碳材料附着于装置而使生产性降低这样的问题。
[0475]
(第3工序)对原料碳材料进行造粒的工序(对原料碳材料进行球形化处理的工序)
[0476]
碳材料优选为通过对原料碳材料赋予冲击压缩、摩擦、剪切力等机械作用而实施了球形化处理(以下,也称为造粒)而得到的碳材料。另外,该球形化石墨优选由多个鳞片状或鳞状石墨、以及被磨碎的石墨微粉形成,特别优选由多个鳞片状石墨形成。
[0477]
本发明优选具有至少赋予冲击、压缩、摩擦及剪切力中的任意的力学能来对原料碳材料进行造粒的造粒工序。作为该工序中使用的装置,可以使用例如反复赋予以冲击力为主体并且也包括原料碳材料的相互作用在内的压缩、摩擦、剪切力等机械作用的装置。
[0478]
具体来说,优选如下的装置:在壳体内部具有设置了多个叶片的转子,并通过该转子高速旋转对导入到内部的原料碳材料赋予冲击、压缩、摩擦、剪切力等机械作用,由此来进行表面处理的装置。另外,优选具有通过使原料碳材料循环而反复赋予机械作用的机构的装置。
[0479]
作为这样的装置,例如可以举出混合系统(hybridizationsystem)(株式会社奈良机械制作制)、kryptron、kryptronorb(earthtechnica公司制)、cf磨(宇部兴产株式会社制)、机械熔融系统、nobilta、faculty(hosokawa micron公司制)、theta composer(株式会社德寿工作所制造)、composi(日本焦炭工业制)等。这些当中,优选株式会社奈良机械制作所制造的混合系统。
[0480]
在使用上述装置进行处理的情况下,例如,旋转的转子的圆周速度优选为30m/秒以上、更优选为50m/秒以上、进一步优选为60m/秒以上、特别优选为70m/秒以上、最优选为80m/秒以上,并且优选为100m/秒以下。在上述范围内时,可以更有效地进行球形化,同时进行微粉对母材的附着及在母材中的内包。
[0481]
另外,对原料碳材料赋予机械作用的处理虽然能够在仅使原料碳材料通过时来进行处理,但优选使原料碳材料装置内循环或滞留30秒钟以上来进行处理,更优选使原料碳材料装置内循环或滞留1分钟以上、进一步优选使原料碳材料装置内循环或滞留3分钟以上、特别优选使原料碳材料装置内循环或滞留5分钟以上来进行处理。
[0482]
另外,造粒剂在上述将原料碳材料和造粒剂混合时的粘度优选为1cp以上、更优选为5cp以上、进一步优选为10cp以上、特别优选为20cp以上,另一方面,优选为1000cp以下、更优选为800cp以下、进一步优选为600cp以下、特别优选为500cp以下。如果粘度在上述范围内,则在造粒材料存在下对原料碳材料进行造粒时,能够防止由于与转子、壳体的碰撞等的冲击力而引起的附着粒子的脱离,另外,造粒材料进入到1nm至4nm的微孔中,在后工序中成为非晶质碳,由此,可以减少该微孔,从而能够制造低温输入输出特性、高温保存特性优异的负极材料,因此优选。上述的在对原料碳材料进行造粒的工序中的粘度可以通过后述的有机溶剂的添加、混合温度的控制来进行调整。
[0483]
另外,在对原料碳材料进行造粒的工序中,也可以在其它物质存在下对原料碳材料进行造粒,作为其它物质,可列举例如:能够与锂合金化的金属或其氧化物、鳞片状石墨、鳞状石墨、被磨碎的石墨微粉、非晶质碳及未焙烧的焦炭等。通过同时使用原料碳材料以外的物质来进行造粒,可以制造各种类型粒子结构的非水系二次电池用碳材料。
[0484]
另外,原料碳材料、造粒剂及上述其它物质可以将全部量一次性投入到上述装置内,也可以分开依次地投入,还可以连续地投入。另外,原料碳材料、造粒剂及上述其它物质可以同时投入到上述装置内,也可以混合后投入,还可以分别投入。原料碳材料、造粒剂和上述其它物质可以同时混合,也可以将上述其它物质添加到原料碳材料和造粒剂混合而得到的混合物中,还可以将原料碳材料添加到其它物质和造粒剂混合而得到的混合物中。根据粒子设计,可以在适当的时期另行添加、混合。
[0485]
在碳材料的球形化处理时,更优选一边使球形化处理中生成的微粉有效地附着于粒子表面一边进行球形化处理。通过一边使球形化处理中生成的微粉附着于粒子表面一边进行球形化处理,小的鳞片致密地粘结于大的鳞片的间隙,能够使鳞片间强固地粘结。因此,在其后用非晶质物或石墨化物包覆时,能够更有效地减少粒子内空隙。另外,能够作为
li离子的插入/脱离位点利用的边缘的量增加、且可以使电解液有效且高效地遍布于粒子内空隙,从而有效地利用粒子内的li离子插入脱离位点,从而存在显示良好的低温输出特性及循环特性的倾向。另外,附着于母材的微粉不限于在球形化处理中生成的微粉、既可以在调整鳞片状石墨粒度时调整为同时包含微粉,也可以在适当的时期另行添加、混合。
[0486]
为了使微粉有效地附着于母材的粒子表面,优选增强鳞片状石墨粒子
‑
鳞片状石墨粒子间、鳞片状石墨粒子
‑
微粉粒子间、及微粉粒子
‑
微粉粒子间的附着力。作为粒子间的附着力,具体可列举:中间不夹有粒子间夹杂物的范德华力及静电引力、中间夹有粒子间夹杂物的物理或化学交联力等。
[0487]
范德华力以平均粒径(d50)100μm为界越小越表现出[自重]<[附着力]。因此,作为球形化石墨原料的鳞片状石墨(原料碳材料)的平均粒径(d50)越小,粒子间附着力越增大,越容易成为微粉附着于母材、以及微粉内包于球形化粒子中的状态,故优选。鳞片状石墨的平均粒径(d50)优选为1μm以上、更优选为2μm以上、进一步优选为3μm以上,且优选为80μm以下、更优选为50μm以下、进一步优选为35μm以下、特别优选为20μm以下、尤其优选为10μm以下、最优选为8μm以下。
[0488]
作为中间夹有粒子间夹杂物的物理或化学交联力,可列举出中间夹有液体性夹杂物、固体性夹杂物的物理或化学交联力。作为上述化学交联力,可列举出在粒子和粒子间夹杂物之间通过化学反应、烧结、力学化学效果等而形成了共价键、离子键、氢键等时的交联力。
[0489]
(第4工序)除去造粒剂的一部分及有机溶剂的工序
[0490]
在本发明中,优选具有前述的除去造粒剂的一部分及有机溶剂的工序。作为除去造粒剂的一部分及有机溶剂的方法,可列举例如:利用溶剂进行清洗的方法、通过在常压、减压下施加热而使造粒剂的一部分及有机溶剂挥发、分解除去的方法。
[0491]
此时的热处理温度优选为60℃以上、更优选为100℃以上、进一步优选为200℃以上、特别优选为300℃以上,且优选为1500℃以下、更优选为1000℃以下、进一步优选为800℃以下、最优选为造粒材的沸点附近。热处理温度在上述范围内时,可以使造粒剂充分地挥发、分解除去,从而削减生产成本,并且成型处理时的成型状态也变得良好,因此优选。
[0492]
热处理时间优选为0.5~48小时、更优选为1~40小时、进一步优选为2~30小时、特别优选为3~24小时。热处理时间在上述范围内时,可以使造粒剂充分地挥发、分解除去,从而可以提高生产性。
[0493]
热处理的气氛可列举大气气氛等活性气氛、或者氮气氛、氩气氛等非活性气氛,在200℃~300℃进行热处理时没有特别限制,但在300℃以上进行热处理时,从防止石墨表面的氧化的观点出发,优选氮气氛、氩气氛等非活性气氛。
[0494]
(第5工序)在造粒碳材料中添加附着结晶性低于原料碳材料的碳物质的工序
[0495]
在本发明中,可以具有在造粒碳材料中进一步添加附着结晶性低于原料碳材料的碳物质的工序。根据该工序,可得到在表面的至少一部分具有非晶质碳物质的石墨,因此,使用该石墨的非水二次电池用负极与电解液的副反应少,可以得到高容量、低温输入输出特性、高温保存特性优异的非水二次电池用负极材料。
[0496]
对造粒碳材料添加附着(复合化)非晶质碳物质的处理是将作为非晶质碳物质的有机化合物与造粒碳材料混合、在非氧化性气氛下、优选为氮、氩、二氧化碳等的流通下进
行加热,从而使有机化合物非晶质碳化的处理。作为成为非晶质碳物质的具体的有机化合物,可以使用石油系、煤系的重油、焦油、沥青,具体可以使用软质或硬质的各种煤焦油沥青、煤液化油等碳系重油、原油的常压或减压蒸馏残渣油等石油系重油、石脑油裂解产生的作为乙烯制造的副产物的裂解系重油等各种物质。
[0497]
另外,可列举出通过对裂解系重油进行热处理而得到的乙烯焦油沥青、fcc滗析油、ashland(
アシュランド
)沥青等热处理沥青等。此外,可列举出聚氯乙烯、聚乙酸乙烯酯、聚乙烯醇缩丁醛、聚乙烯醇等乙烯基类高分子和3
‑
甲基酚醛树脂、3,5
‑
二甲基酚醛树脂等取代酚醛树脂、苊、十环烯、蒽等芳香烃、吩嗪、吖啶等氮环化合物、噻吩等硫环化合物等。另外,作为以固相进行碳化的有机化合物,可列举出纤维素等天然高分子、聚偏氯乙烯、聚丙烯腈等链状乙烯基树脂、聚苯等芳香族类聚合物;糠醇树脂、酚醛树脂、聚酰亚胺树脂等热固性树脂;糠醇这样的热固性树脂原料等。这些当中,优选qi、ti少、在烧成的升温时完全软化的作为软碳的石油系、煤系的重油、焦油、沥青,这是由于,它们在作为非晶质碳时不易生成微孔、能够更有效地减少1nm至4nm的微孔。
[0498]
作为将造粒碳材料和成为非晶质碳物质的有机化合物混合的方法,可列举例如:使用混合器、捏合机将原料碳材料和造粒剂进行混合的方法、将使有机化合物溶解于低粘度稀释溶剂(有机溶剂)而得到的造粒剂与原料碳材料进行混合,然后再除去该稀释溶剂(有机溶剂)的方法等。这些方法中,优选使用混合器、捏合机将原料碳材料和造粒剂进行混合的方法,这是由于,该方法中,有机化合物中的成为碳物质的成分越多,越是能够更有效地减少1nm至4nm的微孔。
[0499]
另外,在将造粒碳材料与作为碳物质的有机化合物混合时,成为碳物质的有机化合物的粘度优选为1cp以上且1000cp以下、更优选为5cp以上且800cp以下、进一步优选为10cp以上且600cp以下、特别优选为20cp以上且500cp以下。粘度为上述范围内时,作为碳物质的有机化合物进入到造粒碳材料的1nm至4nm的微孔中、通过烧成而形成为非晶质碳,由此可以减少该微孔,从而能够制造低温输入输出特性、高温保存特性优异的负极材料,因此优选。
[0500]
将造粒碳材料与作为碳物质的有机化合物混合时的混合温度通常为作为碳物质的有机化合物的软化点以上、优选比软化点高10℃以上的温度、更优选比软化点高20℃以上的温度、进一步优选高30℃以上的温度、特别优选高50℃以上的温度,且通常在450℃以下、优选在250℃以下进行。加热温度过低时,作为碳物质前体的有机化合物的粘度增高,混合变得困难,存在包覆形态变得不均匀的担心,加热温度过高时,由于作为碳物质前体的有机化合物的挥发和缩聚而使混合体系的粘度变高,混合变得困难,存在包覆形态变得不均匀的担心。
[0501]
加热温度(烧成温度)根据有用制备混合物的有机化合物而不同,通常加热至500℃以上、优选加热至600℃以上、更优选加热至700℃以上而在石墨表面的至少一部分添加附着非晶质碳。加热温度的上限是有机化合物的炭化物未达到与混合物中的鳞片状石墨的结晶结构同等的结晶结构的温度,加热温度的上限通常为2000℃、优选为1800℃、更优选为1700℃、进一步优选为1600℃。
[0502]
进行了上述这样的处理后,接下来可以通过适宜地组合实施破碎、粉碎及分级处理来制成碳物质复合碳材料。另外,形状为任意形状,但平均粒径通常为2~50μm、优选为5
~35μm、特别优选为8~30μm。
[0503]
另外,若使用公知的技术,则可以使用能够控制在所要求的范围内的公知的技术。即,可以通过使用公知的技术进行球形化处理来制造经过了球形化的石墨粒子。例如可列举使用反复对粒子赋予以冲击力为主体并且也包括粒子的相互作用在内的压缩、摩擦、剪切力等机械作用的装置来进行。具体来说,优选如下的装置:在壳体内部具有设置了多个叶片的转子,并通过该转子高速旋转对导入到内部的碳材料赋予冲击、压缩、摩擦、剪切力等机械作用,由此来进行表面处理的装置。另外,优选具有通过使石墨粒子循环而反复赋予机械作用的机构的装置。作为具体的装置,可列举例如:混合系统(hybridizationsystem)(株式会社奈良机械制作制)、kryptron(earthtechnica公司制)、cf磨(宇部兴产株式会社制)、机械熔融系统(hosokawa micron公司制)、theta composer(株式会社德寿工作所制)等。这些当中,优选株式会社奈良机械制作所制造的混合系统。例如在使用前述的装置进行处理的情况下,旋转的转子的圆周速度没有特别限制,优选设为30m/秒~100m/秒、更优选设为40m/秒~100m/秒、进一步优选设为50m/秒~100m/秒。另外,处理虽然能够仅使碳物质通过来进行,但优选使其在装置内循环或滞留30秒钟以上来进行处理,更优选使其在装置内循环或滞留1分钟以上来进行处理。
[0504]
<c2
‑
2.石墨的加压处理>
[0505]
本发明的一个实施方式的负极材料的制造优选包含对石墨进行加压处理的工序。作为石墨的加压处理的方法,只要是能够进行加压的方法就没有特别限制,可列举例如:将原料石墨加入到橡胶型等的容器中、以水作为加压介质的静水压各向同性加压处理、以及以空气等气体作为加压介质的基于气压的各向同性加压处理。另外,也可以将原料石墨填充至模具中以单向加压沿恒定方向进行加压处理
[0506]
进行加压的时间可以在制造上述球形化石墨的工序1~工序5的任意工序中实施,但在工序4与5之间实施由于是在除去了多余的造粒材料的状态下进行加压,可更有效地施加压力,因此优选。
[0507]
作为石墨的加压处理的加压介质的压力,通常优选为100mpa以上、更优选为150mpa以上、进一步优选为200mpa以上,其上限通常为400mpa以下、优选为300mpa以下。为上述下限值以下时,基于成型的微孔的控制变得不充分,每单位覆盖率的粒子内孔容难以达到优选的范围,为上述上限值以上时,装置变得过大,过度损害量产性,从这样的观点出发,不优选。
[0508]
成型处理后的样品优选进行一次粉碎、破碎、粉碎处理至与处理前的粒径同等或其以下。其中,作为粉碎的方法,可以使用公知的装置,成型体大的情况下,优选经过粗粉碎工序、中间粉碎工序、粉碎工序这样的工序来实施。作为粗粉碎工序的装置,可列举例如:颚式破碎机、冲击式压碎机、锥形压碎机、旋转复合机等,作为中间粉碎机,可列举辊式压碎机、定向磨、锤磨机等,作为微粉碎设备,可列举涡轮磨、cryptron、销棒粉碎机、球磨、振动磨、粉碎机等。上述粉碎之后,实施分级处理,粉碎至与原料同等以下。比处理前的粒径大的情况下,粒子彼此间通过成型处理而显示出附着凝聚,在非晶质包覆时无法在该部分良好地包覆,因此不优选。另一方面,在明显小于处理前的粒径的情况下,将球形化石墨进行了粉碎而导致sa提高,因此不优选。
[0509]
(累积孔容)
[0510]
本发明的一个实施方式的负极材料的原料所使用的石墨优选在使用压汞法(汞孔隙率法)测定的微孔分布中具有2个以上的峰,对于极小值以下的范围的累积孔容而言,以使用压汞法(汞孔隙率法)测定的值计,通常为0.15ml/g以下、优选为0.08ml/g以下、更优选为0.070ml/g以下、进一步优选为0.060ml/g以下、特别优选为0.050ml/g以下。累积孔容大于上述范围的情况下,表示粒子内空隙大,用非晶质包覆时难以有效地降低孔容,因此不优选。
[0511]
(较小一侧的峰顶)
[0512]
本发明的一个实施方式的负极材料的原料所使用的石墨优选在使用压汞法(汞孔隙率法)测定的微孔分布中具有2个以上的峰,对于微孔径较小的一侧的峰的峰顶而言,以使用压汞法(汞孔隙率法)测定的值计,通常为500nm以下、优选为400nm以下、更优选为300nm以下、进一步优选为200nm以下、特别优选为100nm以下。粒子内空隙大于上述范围的情况下,在进行了球形化的时刻,作为原料的鳞片状石墨彼此间的距离远,存在较大的空隙,因此,在用非晶质包覆时无法有效地粘结,因此不优选。
[0513]
(圆度)
[0514]
本发明的一个实施方式的负极材料原料所使用的石墨的通过流式粒子图像分析求出的圆度优选为0.88以上、更优选为0.90以上、进一步优选为0.91以上、特别优选为0.92以上、最优选为0.93以上。如果是这样的圆度高的石磨,则使用其制造的负极材料的li离子扩散的弯曲度下降而使粒子间空隙中的电解液移动变得顺利,从而可以提高快速充放电特性,因此优选。另一方面,该圆度的理论上限为1,因此通常小于1、优选为0.99以下、更优选为0.98以下、进一步优选为0.97以下。从确保所得到的负极材料彼此的接触性而提高循环特性的观点出发,优选为上述上限以下。
[0515]
圆度是使用流式粒子图像分析装置(东亚医疗电子株式会社制fpia
‑
2000)、进行基于等效圆直径的粒径分布的测定并通过计算出平均圆度而求得。圆度用以下的式子来定义,圆度为1时成为理论上的真球。
[0516]
[圆度]=[具有与粒子投影形状相同面积的等效圆的周长]/[粒子投影形状的实际周长]
[0517]
在该圆度的测定中,作为分散介质,使用了离子交换水,作为表面活性剂,使用了聚氧乙烯(20)单月桂酸酯。等效圆直径是指具有与拍摄的粒子图像相同投影面积的圆(等效圆)的直径,圆度是指,以等效圆的周长为分子、以拍摄的粒子投影图像的周长为分母而得到的比率。将测得的等效直径为10~40μm范围的粒子的圆度取平均,作为圆度。
[0518]
(振实密度)
[0519]
石墨的振实密度优选为0.60g/cm3以上、更优选为0.70g/cm3以上、进一步优选为0.80g/cm3以上、特别优选为0.855/cm3以上、最优选为0.90g/cm3以上,另一方面,通常为1.40g/cm3以下、优选为1.30g/cm3以下、更优选为1.20g/cm3以下。
[0520]
(体积基准平均粒径)
[0521]
石墨的体积基准平均粒径(d50)没有特别限定,通常为1μm以上、优选为3μm以上、更优选为5μm以上,且通常为50μm以下、优选为40μm以下、更优选为30μm以下。
[0522]
(bet比表面积)
[0523]
石墨的bet比表面积没有特别限定,通常为1.0m2/g以上、优选为1.5m2/g以上、更优
选为2.0m2/g以上、进一步优选为3.0m2/g以上、特别优选为4.5m2/g以上、最优选为5.0m2/g以上,另外,通常为30.0m2/g以下、优选为20.0m2/g以下、更优选为10.0m2/g以下。
[0524]
(拉曼r1值)
[0525]
石墨的由下述式α表示的拉曼r1值没有特别限定,优选为0.10以上且1.00以下。另外,拉曼r值更优选为0.15以上、进一步优选为0.20以上、特别优选为0.25以上,另一方面,更优选为0.80以下、进一步优选为0.60以下。
[0526]
式α:
[0527]
拉曼r1值=(拉曼光谱分析中1360cm
‑1附近的峰p
b
的强度i
b
)/(1580cm
‑1附近的峰p
a
的强度i
a
)
[0528]
<c2
‑
3.石墨的非晶质碳复合化>
[0529]
本发明的一个实施方式的负极材料包含在表面的至少一部分具有非晶质碳物质或石墨质物质中的至少一者的石墨。这里,非晶质碳物质是指,基于x射线广角衍射法的(002)面的面间距(d002)通常为0.340nm以上的碳,非晶质碳物质与碳物质含义相同。另一方面,石墨质物质是指(d002)低于0.340nm的石墨。该非晶质碳复合化的处理通过将石墨与非晶质碳前体(非晶质碳物质的原料)混合并进行烧成来进行。通过在适宜的条件下进行石墨的非晶质碳复合化,在实现满足前述的热特性的同时,孔容也容易控制在前述的范围。
[0530]
作为非晶质碳前体,没有特别限定,可列举:焦油及沥青、萘、蒽及其衍生物等芳香烃类、酚醛树脂、聚乙烯醇等热塑性高分子等有机物。这些有机物前体可以仅使用一种,也可以组合2种以上使用。这些当中,从碳结构容易发达、能够以较少的量进行包覆这样的观点出发,优选焦油、沥青、芳香烃类,为了满足前述的覆盖率与微孔量的关系,优选残碳为50%以上者,更优选残碳为60%、特别优选残碳为65%以上。残碳率越高,实施烧成处理时的发泡、体积减少越是被抑制,从而能够效率良好地填埋上述石墨中的粒子内微孔,因此优选。
[0531]
本发明的非晶质碳前体中所含的金属杂质量定义为:将相对于非晶质碳前体的总重量的fe、al、si、ca的总含量除以残碳率而得到的值。相对于非晶质碳前体的总重量优选为1000重量ppm以下、更优选为400重量ppm以下、进一步优选为100重量ppm以下、特别优选为50重量ppm以下,且通常为0.1重量ppm以上。如果非晶质碳前体中所含的金属杂质量为上述范围内,则容易满足前述的覆盖率与微孔量的关系,因此优选。
[0532]
非晶质碳前体中所含的喹啉不溶成分(qi)相对于非晶质碳前体的总重量优选为5重量%以下、更优选为2重量%以下、进一步优选为1重量%以下、特别优选为0.5重量%以下、最优选为0.1重量%以下。如果非晶质碳前体中所含的喹啉不溶成分(qi)为上述范围内,则容易满足前述的覆盖率与微孔量的关系,因此优选。
[0533]
将石墨与非晶质碳前体混合后,进行烧成。进行烧成时的温度优选为950℃以上、更优选为1000℃以上、进一步优选为1050℃以上、特别优选为1100℃以上、最优选为1150℃以上,另一方面,优选为2000℃以下、更优选为1800℃以下、进一步优选为1600℃以下、特别优选为1500℃以下。另外,进行烧成时的时间优选为0.5小时以上、更优选为1小时以上,另一方面,优选为1000小时以下、更优选为500小时以下、进一步优选为100小时以下。如果进行烧成时的温度、时间为上述范围,则容易满足前述的覆盖率与微孔量的关系,因此优选。
[0534]
进行烧成时的气氛优选为非活性气氛下。具体可列举:通过使氮、氩这样的非活性
气体流通来降低氧浓度的方法、通过减压处理将氧排出至体系外并用氮、氩来恢复压力的方法、通过将焦炭粉等牺牲材料填充至制品的周围来降低炉内气氛中所含的氧的方法。可以通过非活性气体的流通量、流通时间、减压处理的程度、牺牲材料的填充条件等来控制体系内的氧浓度。氧浓度(体积浓度)优选为3%以下、更优选为1%以下、进一步优选为1000ppm以下、特别优选为500ppm以下、最优选为100ppm以下。如果超过上述范围,则非晶质碳物质的碳结构的发达受到阻碍,存在非晶质本身产生微孔而难以满足前述的覆盖率与微孔量的关系的倾向。
[0535]
在石墨中混合非晶质碳前体时的非晶质碳前体的混合比率应该基于目标的复合粒子的组成适宜选择,对于相对于石墨的非晶质碳前体的量而言,以作为其残炭物(非晶质碳)的重量比(〔[非晶质碳的重量]/[石墨的重量]〕
×
100)计,通常为0.01%以上、优选为0.1%以上、更优选为0.5%以上、进一步优选为1%以上、特别优选为2%以上、最优选为3%以上,另一方面,优选为30%以下、更优选为20%以下、进一步优选为15%以下、特别优选为10%以下。如果该重量比为上述范围,则电池为高容量、且li离子容易插入/脱离,因此低温输入输出特性、快速充放电特性及循环特性优异,从这点来看是优选的。
[0536]
<石墨的石墨质物质复合化>
[0537]
本发明的一个实施方式的负极材料包含在表面的至少一部分具有非晶质碳物质或石墨质物质中的至少一者的石墨。该石墨化的处理通过将石墨与有机化合物(石墨质物质的原料)混合、烧成来进行。
[0538]
包含在表面的至少一部分具有石墨质物质的石墨的负极材料的制造方法没有特别限制,可以优选利用以下的工序来制造包含在表面的至少一部分具有石墨质物质的石墨的负极材料,所述工序为:对上述的石墨(也称为“原料石墨”)进行加压处理后进行破碎,再与用于获得石墨质物质包覆部分的有机化合物混合,对所得到的混合物进行石墨化、粉碎处理的工序;或者,将原料碳材料与用于获得石墨质物质包覆部分的有机化合物混合并进行加压处理,然后对所得到的混合物进行石墨化、粉碎处理的工序。更优选为粉碎工序数少的后者。
[0539]
作为有机化合物,具体可列举:含浸沥青、煤焦油沥青、煤液化油等煤系重油、沥青烯等直馏系重油、及乙烯重尾焦油等分解系重油等石油系重油等示例出的易石墨化有机化合物、芳香烃、含氮环状化合物、含硫环状化合物、聚苯、聚氯乙烯、聚乙烯醇、聚丙烯腈、聚乙烯醇缩丁醛、天然糠醇高分子、聚苯硫醚、聚苯醚、树脂、酚醛树脂、酰亚胺树脂等,其中,优选能够通过烧成而石墨化或碳化的易石墨化有机化合物。
[0540]
将原料石墨与有机化合物混合的工序/进行加压处理的工序可以按照与石墨的非晶质碳复合化同样的方式来进行。
[0541]
·
对混合物进行烧成的工序
[0542]
具体为以下工序:将混合物在非氧化性气氛下、优选在氮、氩、二氧化碳等的流通下进行加热,使有机化合物石墨化,从而制造包含在表面的至少一部分具有石墨质物质的石墨的非水二次电池用复合负极材料。
[0543]
烧成温度根据用于制备混合物的有机化合物而不同,在得到石墨质物质被复合化了的非水二次电池用负极材料的情况下,只要为有机化合物进行石墨化的温度或其以上的温度即可,具体来说,通常加热至2000℃以上、优选加热至2500℃以上、更优选加热至2700
℃以上来使其充分地炭化。加热温度的上限为有机化合物的炭化物未达到与混合物中的碳材料的结晶结构同等的结晶结构的温度,通常为3300℃以下、优选为3100℃以下、更优选为3000℃以下。
[0544]
在烧成处理条件中,热历程温度条件、升温速度、冷却速度、热处理时间等可适宜设定。另外,可以在比较低温的区域进行了热处理后、升温至给定的温度。需要说明的是,本工序中使用的反应机可以是间歇式,也可以是连续式,另外,可以为一台,也可以为多台。
[0545]
烧成所使用的炉只要满足上述要件就没有特别限制,例如可以使用梭式炉、隧道炉、里德哈默炉、旋转窑炉、高压釜等的反应槽、焦化器(焦炭制造的热处理槽)、坦曼式炉、艾奇逊炉,加热方式可以采用高频感应加热炉、直接式电阻加热、间接式电阻加热、直接燃烧加热、辐射加热等。处理时可以根据需要进行搅拌。
[0546]
经过了上述工序的复合碳材料根据需要可进行再次粉碎、破碎、分级处理等粉体加工,从而得到非水二次电池用负极材料。
[0547]
用于粉碎、破碎的装置没有特别限定,例如,作为粗粉碎机,可列举剪切式磨、颚式破碎机、冲击式压碎机、锥形压碎机等,作为中间粉碎机,可列举辊式压碎机、锤磨机等,作为微粉碎机,可列举球磨、振动磨、销棒粉碎机、搅拌磨、喷射磨等。
[0548]
作为用于分级处理的装置,没有特别限制,例如,在干式筛分的情况下,可以使用旋转式筛、摇动式筛、转动式筛、振动式筛等,在干式气流式分级的情况下,可以使用重力式分级机、惯性力式分级机、离心力式分级机(分粒器、旋风分离器)等,另外,在湿式筛分的情况下,可以使用机械式湿式分级机、水力分级机、沉降分级机、离心式湿式分级机等。
[0549]
在石墨中混合有机化合物时的有机化合物的混合比率应该基于目标的复合粒子的组成适宜选择,对于相对于石墨的有机化合物的量而言,以作为其残炭物(石墨质物质)的重量比(〔[石墨质物质的重量]/[具有石墨质物质的石墨]〕
×
100)计,通常为0.01%以上、优选为0.1%以上、更优选为0.5%以上、进一步优选为1%以上、特别优选为2%以上、最优选为3%以上,另一方面,优选为30%以下、更优选为20%以下、进一步优选为15%以下、特别优选为10%以下。如果该重量比为上述范围,则电池为高容量、且li离子容易插入/脱离,因此低温输入输出特性、快速充放电特性及循环特性优异,从这点来看是优选的。
[0550]
<c2
‑
4.其它处理>
[0551]
为了制造本发明的一个实施方式的负极材料,可以对通过前述的制造方法得到的复合粒子另行进行粉碎处理。
[0552]
作为粉碎处理所使用的粗粉碎机,可列举颚式破碎机、冲击式压碎机、锥形压碎机等,作为中间粉碎机,可列举辊式压碎机、定向磨、锤磨机等,作为微粉碎设备,可列举涡轮磨、cryptron、球磨、振动磨、搅拌磨、喷射磨等。这些当中,从粉碎时间短、处理速度的观点出发,优选涡轮磨、cryptron。
[0553]
为了制造本发明的一个实施方式的负极材料,可以对通过前述的制造方法得到的复合粒子进行粒径的分级处理。作为分级处理条件,可以使用开孔通常为53μm以下、优选为45μm以下、更优选为38μm以下的装置来实施。
[0554]
作为用于分级处理的装置,没有特别限制,例如,在干式筛分的情况下,可以使用旋转式筛、摇动式筛、转动式筛、振动式筛等,在干式气流式分级的情况下,可以使用重力式分级机、惯性力式分级机、离心力式分级机(分粒器、旋风分离器)等,在湿式筛分的情况下,
可以使用机械式湿式分级机、水力分级机、沉降分级机、离心式湿式分级机等。
[0555]
〔c3.非水二次电池用负极〕
[0556]
本发明的一个实施方式的非水二次电池用负极(以下,有时称为“本发明的负极”)具备:集电体、和形成在该集电体上的活性物质层,其中,该活性物质层含有上述的本发明的一个实施方式的负极材料。本实施方式的非水二次电池用负极除了活性物质层以外,可以与第1实施方式同样。
[0557]
使用上述负极材料制作负极时,利用水性或有机类介质将在负极材料中配合有粘结树脂的材料制成浆料,并根据需要而向其中加入增稠剂后涂布在集电体、进行干燥即可。
[0558]
作为粘结树脂,优选使用相对于非水电解液稳定、且非水溶性的树脂。例如可使用:丁苯橡胶、异戊二烯橡胶及乙丙橡胶等橡胶状高分子;聚乙烯、聚丙烯、聚对苯二甲酸乙二醇酯、聚酰亚胺、聚丙烯酸、及芳香族聚酰胺等合成树脂;苯乙烯
‑
丁二烯
‑
苯乙烯嵌段共聚物或其加氢产物、苯乙烯
‑
乙烯
‑
丁二烯、苯乙烯共聚物、苯乙烯
‑
异戊二烯及苯乙烯嵌段共聚物及其氢化物等热塑性弹性体;间规立构
‑
1,2
‑
聚丁二烯、乙烯
‑
乙酸乙烯酯共聚物、及乙烯与碳原子数3~12的α
‑
烯烃的共聚物等软质树脂状高分子;四氟乙烯
‑
乙烯共聚物、聚偏氟乙烯、聚五氟丙烯及聚六氟丙烯等氟化高分子等。作为有机类介质,可使用例如n
‑
甲基吡咯烷酮及二甲基甲酰胺。
[0559]
相对于负极材料100重量份,通常使用0.1重量份以上、优选使用0.2重量份以上的粘结树脂。通过使粘结树脂的使用量相对于负极材料100重量份为0.1重量份以上,负极材料相互间的粘结力、负极材料与集电体的粘结力变得充分,可以防止由于从负极剥离负极材料而引起的电池容量的减少及循环特性的变差。
[0560]
另外,粘结树脂的使用量相对于负极材料100重量份优选为10重量份以下、更优选为7重量份以下。通过使粘结树脂的使用量相对于负极材料100重量份为10重量份以下,可以防止负极容量的减少、且防止锂离子等碱金属离子相对于负极材料的出入受到阻碍等问题。
[0561]
作为添加到浆料中的增稠剂,可列举例如:羧甲基纤维素、甲基纤维素、羟乙基纤维素及羟丙基纤维素等水溶性纤维素类、聚乙烯醇及聚乙二醇等。这些当中,优选羧甲基纤维素。优选以相对于负极材料100重量份通常为0.1~10重量份、特别是0.2~7重量份来使用增稠剂。
[0562]
作为负极集电体,使用目前已知可用于该用途的例如铜、铜合金、不锈钢、镍、钛及碳等即可。集电体的形状通常为片状,还优选使用其表面带有凹凸的材料、网状物及冲孔金属等。
[0563]
在集电体上涂布负极材料和粘结树脂的浆料并进行干燥之后,优选进行加压而使形成在集电体上的活性物质层的密度增大、从而使负极活性物质层的每单位体积的电池容量增大。活性物质层的密度优选在1.2~1.8g/cm3的范围、更优选为1.3~1.6g/cm3。通过使活性物质层的密度为上述下限值以上,可以防止伴随着电极厚度的增大而带来的电池容量的降低。另外,通过使活性物质层的密度为上述上限值以下,伴随电极内的粒子间空隙减少而导致空隙中所保持的电解液量减少,可以防止锂离子等碱金属离子的移动性变小而使快速充放电特性降低。
[0564]
〔c3.非水二次电池〕
[0565]
本发明的一个实施方式的非水二次电池是具备正极、负极、以及电解质的非水二次电池,其中,作为负极,使用的是本发明的一个实施方式的负极。特别是,本发明的一个实施方式的非水二次电池中使用的正极及负极通常优选为能够吸留及放出li离子的正极及负极,本发明的一个实施方式的非水二次电池优选为锂离子二次电池。
[0566]
本发明的一个实施方式的非水二次电池除了使用了上述的本发明的一个实施方式的负极以外,可以按照常规方法来制造。特别是,本发明的一个实施方式的非水二次电池优选将[负极的容量]/[正极的容量]的值设计成1.01~1.5、更优选设计成1.2~1.4。
[0567]
[c3
‑
1.正极]
[0568]
作为本发明的一个实施方式的非水二次电池的成为正极的活性物质的正极材料,例如使用基本组成以licoo2表示的锂钴复合氧化物、以linio2表示的锂镍复合氧化物、以limno2及limn2o4表示的锂锰复合氧化物等锂过渡金属复合氧化物、二氧化锰等过渡金属氧化物、以及这些复合氧化物的混合物等即可。进一步,可使用tis2、fes2、nb3s4、mo3s4、cos2、v2o5、cro3、v3o3、feo2、geo2及lini
0.33
mn
0.33
co
0.33
o2、lifepo4等。
[0569]
可以通过将在上述正极材料中配合有粘结树脂的材料利用适当的溶剂进行浆料化并涂布在集电体上、并进行干燥而制作正极。需要说明的是,在浆料中,优选含有乙炔黑及科琴黑等导电材料。另外,也可以根据需要而含有增稠剂。需要说明的是,作为粘结材料及增稠剂,可以使用其用途公知的那些,例如可以使用作为用于负极的制造而示例出的那些。
[0570]
相对于正极材料100重量份,导电材料的配合量优选为0.5重量份~20重量份、更优选为1重量份~15重量份。另外,相对于正极材料100重量份,增稠剂的配合量优选为0.2重量份~10重量份、更优选为0.5重量份~7重量份。此外,就相对于正极材料100重量份的粘结树脂的配合量而言,在用水将粘结树脂浆料化的情况下,优选为0.2重量份~10重量份、更优选为0.5重量份~7重量份,另一方面,在用n
‑
甲基吡咯烷酮等溶解粘结树脂的有机溶剂将粘结树脂浆料化的情况下,优选为0.5重量份~20重量份、更优选为1~15重量份。
[0571]
作为正极集电体,可列举例如:铝、钛、锆、铪、铌及钽等、以及它们的合金。这些当中,优选铝、钛及钽、及其合金,最优选为铝及其合金。
[0572]
[c3
‑
2.电解液]
[0573]
电解液可使用在以往周知的非水溶剂中溶解各种锂盐而成的溶液。
[0574]
作为非水溶剂,可使用例如碳酸亚乙酯、氟代碳酸亚乙酯、碳酸亚丙酯、碳酸亚丁酯及碳酸亚乙烯酯等环状碳酸酯;碳酸二甲酯、碳酸甲乙酯及碳酸二乙酯等链状碳酸酯;γ
‑
丁内酯等环状酯;冠醚、2
‑
甲基四氢呋喃、四氢呋喃、1,2
‑
二甲基四氢呋喃及1,3
‑
二氧戊环等环状醚;1,2
‑
二甲氧基乙烷等链状醚等。通常可以将这些溶剂中的2种以上混合使用。这些当中,优选使用环状碳酸酯和链状碳酸酯,或者在其中进一步混合其它溶剂后使用。
[0575]
也可以在电解液中添加碳酸亚乙烯酯、乙烯基碳酸亚乙酯、琥珀酸酐、马来酸酐、丙磺酸内酯及二乙基砜等化合物、二氟磷酸锂这样的二氟磷酸盐等。进一步,还可以添加二苯基醚及环己基苯等过充电防止剂。
[0576]
作为溶解于非水溶剂的电解质,可列举例如:liclo4、lipf6、libf4、licf3so3、lin(cf3so2)2、lin(cf3cf2so2)2、lin(cf3so2)(c4f9so2)及lic(cf3so2)3等。电解液中的电解质的浓度通常为0.5mol/l~2mol/l、优选为0.6mol/l~1.5mol/l。
[0577]
[c3
‑
3.隔板]
[0578]
本发明的一个实施方式的非水二次电池中优选使用隔板,且该隔板介于正极和负极之间。作为这样的隔板,优选使用聚乙烯、聚丙烯等聚烯烃的多孔性片、或无纺布。
[0579]
实施例
[0580]
[实验a]
[0581]
以下,通过实施例对本发明的内容更具体地进行说明,但本发明只要不超出其主旨,就不受以下的实施例的限定。需要说明的是,以下的实施例中的各种制造条件、评价结果的值具有作为本发明的实施方式中的上限或下限的优选的值的含义,优选的范围也可以是将前述的上限或下限的值与下述实施例的值组合、或者将实施例的值彼此组合而规定的范围。
[0582]
<负极片的制作>
[0583]
使用在后述的条件下制备的负极材料作为负极活性物质,制作了具有活性物质层密度1.6
±
0.03g/cm3(表1的实施例、比较例)、及1.5
±
0.03g/cm3(表2的实施例、比较例)的活性物质层的极板。具体地,在负极材料50.00
±
0.02g中将1重量%羧甲基纤维素钠盐水溶液50.00
±
0.02g(换算成固体成分为0.500g)、及重均分子量27万的苯乙烯
‑
丁二烯橡胶水性分散体1.00
±
0.05g(换算成固体成分为0.5g)用keyence制混合搅拌机搅拌5分钟并脱泡30秒钟,得到了浆料。
[0584]
使用模涂机将该浆料以10cm的宽度涂布在作为集电体的厚度10μm的铜箔上,使得负极材料附着10.00
±
0.2mg/cm2,干燥后,切成宽度5cm,使用直径20cm的辊进行辊压,并调整为活性物质层的密度为1.5
±
0.03g/cm3及1.60
±
0.03g/cm3,得到了各自的电极片。
[0585]
<正极片的制作>
[0586]
如下所述制作了正极。首先,将作为正极活性物质的镍
‑
锰
‑
钴酸锂(linimncoo2)85重量%、作为导电材料的乙炔黑10重量%、作为粘结剂的聚偏氟乙烯(pvdf)5重量%在n
‑
甲基吡咯烷酮溶剂中进行混合,得到了浆料。使用刮板涂布机将该浆料涂布在作为集电体的厚度15μm的铝箔上,使得正极材附着22.5
±
0.2mg/cm2,在130℃下进行了干燥。进一步进行辊压,调整为正极密度达到2.60
±
0.05g/cm3,得到了电极片。
[0587]
<非水二次电池(层压型电池)的制作法>
[0588]
将利用上述方法制作的附着有10.0
±
0.3mg/cm2负极材料、且活性物质层的密度为1.6
±
0.03g/cm3(表1的实施例、比较例)、1.5
±
0.03g/cm3(表2的实施例、比较例)的负极片、正极片、及聚乙烯制隔板按照负极、隔板、正极的顺序进行了层叠。用筒状的铝层压膜包入上述得到的电池元素,注入电解液,该电解液是在碳酸亚乙酯(ec)、碳酸二甲酯(dmc)和碳酸甲乙酯(emc)的混合溶剂(体积比=3:3:4)中以1mol/l溶解有lipf6而得到的,然后进行真空密封,分别制作了片状的非水二次电池。进一步,为了提高电极间的密合性,用玻璃板夹持片状电池进行了加压。
[0589]
<非水二次电池(硬币型电池)的制作法>
[0590]
将利用上述方法制作的附着有10.0
±
0.3mg/cm2负极材料、且活性物质层的密度为1.6
±
0.03g/cm3(表1的实施例、比较例)、1.5
±
0.03g/cm3(表2的实施例、比较例)的电极片冲裁成直径12.5mm的圆盘状,作为负极,将锂金属箔冲裁成直径14mm的圆板状,作为对电极。在两极间放置含浸有电解液的隔板(多孔性聚乙烯膜制),分别制作了2016硬币型电池,
所述电解液是在碳酸亚乙酯和碳酸甲乙酯的混合溶剂(体积比=3:7)中以1mol/l溶解有lipf6而得到的。
[0591]
<放电容量(mah/g)、初始效率(%)>
[0592]
使用利用上述方法制作的非水二次电池(2016硬币型电池),按照下面的测定方法测定了电池充放电时的容量。
[0593]
以0.05c的电流密度对锂对电极充电至5mv,再以5mv的恒定电压进行充电,直至电流密度达到0.005c,将锂掺杂到负极中后,以0.1c的电流密度对锂对电极进行了放电直至1.5v。将此时的放电容量作为本材料的放电容量,将放电容量相对于充电容量之比(放电容量/充电容量
×
100)作为初始效率(%)。
[0594]
<快速放电特性(%)>
[0595]
使用通过前述的非水二次电池的方法制作的层压型非水二次电池,按照下面的测定方法测定了快速放电特性。
[0596]
对于未经充放电循环的非水二次电池,在25℃以电压范围4.1v~3.0v、电流值0.2c(将用1小时将1小时率的放电容量的额定容量放完电的电流值设为1c,下同)进行3个循环,并以电压范围4.2v~3.0v、电流值0.2c(充电时以4.2v进一步实施了恒定电压充电2.5小时)进行2个循环,进行了初始充放电。进而,以电流值0.2c进行恒定电流充电直至4.2v后,再以4.2v进一步实施了2.5小时的恒定电压充电,以3c进行了恒定电流放电直至3.0v。将该3c放电时的放电容量与0.2c放电时的放电容量(上述初始充放电第5循环的放电容量)之比(3c/0.2c
×
100)作为快速放电特性(%)。
[0597]
<低温输出特性>
[0598]
使用通过前述的非水二次电池的方法制作的层压型非水二次电池,按照下面的测定方法测定了低温输出特性。
[0599]
对于未经充放电循环的非水二次电池,在25℃以电压范围4.1v~3.0v、电流值0.2c(将用1小时将1小时率的放电容量的额定容量放完电的电流值设为1c,下同)进行3个循环,并以电压范围4.2v~3.0v、电流值0.2c(充电时以4.2v进一步实施了恒定电压充电2.5小时)进行2个循环,进行了初始充放电。进而,以电流值0.2c进行充电直至soc 50%,然后在
‑
30℃的低温环境下以1/8c、1/4c、1/2c、1.5c、2c的各电流值使其进行恒定电流放电2秒钟,测定在各条件的放电中2秒钟后的电池电压的下降值,由这些测定值计算出充电上限电压为3v时能够在2秒钟流过的电流值i,将用3
×
i(w)这样的式子计算的值作为各个电池的低温输出特性。表1中的低温输出示出的是以比较例a1作为100%时的数值,表2中的低温输出示出的是以比较例a4作为100%时的数值。
[0600]
<高温保存特性(%)>
[0601]
使用通过前述的非水二次电池的方法制作的层压型非水二次电池,按照下面的测定方法测定了高温保存特性
[0602]
对于未经充放电循环的非水二次电池,在25℃以电压范围4.1v~3.0v、电流值0.2c(将用1小时将1小时率的放电容量的额定容量放完电的电流值设为1c,下同)进行3个循环,并以电压范围4.2v~3.0v、电流值0.2c(充电时以4.2v进一步实施了恒定电压充电2.5小时)进行2个循环,进行了初始充放电。进而,以电流值0.2c进行充电直至soc 80%,然后在60℃下进行了2周保存处理。然后,以电流值0.2c进行了放电后,进一步以电流值0.2c
进行充放电(充电时以4.2v进一步实施了恒定电压充电2.5小时),将此时的放电容量作为“保存后的放电容量”。然后,将上述的保存后的放电容量相对于初始充放电第5循环的放电容量之比(保存后的放电容量/初始充放电第5循环的放电容量
×
100(%))作为高温保存特性(%)进行了评价。
[0603]
(实施例a1)
[0604]
将对平均粒径(d50)为100μm的鳞片状天然石墨进行球形化处理而制作的球形化天然石墨(平均粒径(d50)16.2μm、bet比表面积(sa)6.9m2/g、振实密度1.00g/cm3)填充至橡胶制容器中并进行密闭,以200mpa进行各向同性加压处理,由此控制了粉体物性。将得到的成型物进行了破碎、分级处理。将所得到的球形化石墨粉末与作为非晶质碳前体的被调整为灰分<0.01%、金属杂质量60ppm、qi<0.1%的焦油进行混合后,进行减压处理使炉内压力为10torr以下,用氮进行压力恢复直至大气压,进一步流通氮而使炉内的氧浓度<100ppm,实施了1300℃热处理。将所得到的烧成物进行破碎、分级处理,由此得到了负极材料,该负极材料是在石墨粒子的表面含有非晶质碳物质的复合碳材料。由烧成产率确认了所得到的负极材料(复合碳材料)中球形化石墨粒子与非晶质碳的重量比率(球形化石墨质粒子:非晶质碳)为1:0.03。另外,利用前述的测定法测定了差示热分析法(dta)、各拉曼r1~r3值、拉曼半峰宽(δνb)、平均粒径(d50)、bet比表面积(sa)、振实密度、累积孔容、面间距(d002)、微晶尺寸(lc)等负极材料(复合碳材料)的各特性。进一步,利用前述的测定法测定了负极密度1.6g/cm3的电池的放电容量、初始效率、快速放电特性、低温输出特性、高温保存特性等电池特性。将结果示于表1。
[0605]
(实施例a2)
[0606]
除了将球形化石墨粒子与非晶质碳的重量比率(球形化石墨质粒子:非晶质碳)设为1:0.015以外,按照与实施例a1同样的方法得到了负极材料。将与实施例a1进行了同样的测定的结果示于表1。
[0607]
(比较例a1)
[0608]
除了未进行各向同性加压处理、且使氮流通直至炉内的氧浓度<
[0609]
1000ppm而实施了1300℃热处理以外,按照与实施例a1同样的方法得到了负极材料。与实施例a1进行了同样的测定,将其结果示于表1。
[0610]
(比较例a2)
[0611]
除了在非活性气体中实施了900℃热处理以外,按照与实施例a1同样的方法得到了负极材料。与实施例a1进行了同样的测定,将其结果示于表1。
[0612]
(比较例a3)
[0613]
除了在非活性气体中实施了900℃热处理以外,按照与比较例a1同样的方法得到了复合碳材料。与实施例a1进行了同样的测定,将其结果示于表1。
[0614]
(参考例a1)
[0615]
直接使用将平均粒径(d50)为100μm的鳞片状天然石墨进行球形化处理而制作的球形化天然石墨(平均粒径(d50)16.2μm、sa 6.9m2/g、振实密度1.00g/cm3),与实施例a1进行了同样的测定,将其结果示于表1。
[0616]
(实施例a3)
[0617]
将平均粒径(d50)为100μm的鳞片状天然石墨进行粉碎,得到了d50为8.5μm的鳞片
状天然石墨。在所得到的鳞片状天然石墨中混合作为造粒剂的液态油12重量份后,进行球形化处理,进而通过热处理除去造粒材料,得到了球形化天然石墨(平均粒径(d50)12.6μm、sa 19.1m2/g、振实密度0.94g/cm3)。将所得到的球形化石墨填充至橡胶制容器中并进行密闭,以200mpa进行各向同性加压处理,由此控制了粉体物性。将得到的成型物进行了破碎、分级处理。将所得到的球形化石墨粉末与作为非晶质碳前体的被调整为灰分0.02重量%、金属杂质量20重量ppm、qi 1重量%的沥青进行混合,进行减压处理使炉内压力为10torr以下,用氮进行压力恢复直至大气压,进一步流通氮而使炉内的氧浓度<100ppm,在非活性气体中实施了1300℃热处理。将所得到的烧成物进行破碎、分级处理,由此得到了在石墨粒子的表面含有非晶质碳物质的复合碳材料。由烧成产率确认了所得到的复合碳材料中球形化石墨粒子与非晶质碳的重量比率(球形化石墨质粒子:非晶质碳)为1:0.06。除了将负极密度设为1.5g/cm3以外,进行了与实施例a1同样的电池特性的测定,将其结果示于表2。
[0618]
(实施例a4)
[0619]
除了将球形化石墨粒子与非晶质碳的重量比率(球形化石墨质粒子:非晶质碳)设为1:0.08以外,按照与实施例a1同样的方法得到了复合碳材料。进行了与实施例a3同样的测定,将其结果示于表2。
[0620]
(比较例a4)
[0621]
将平均粒径(d50)为100μm的鳞片状天然石墨粉碎,得到了d50为8.5μm的鳞片状天然石墨。通过加热使作为造粒剂的沥青成为液状而混合到所得到的鳞片状天然石墨中,然后同样地边加热边进行了球形化处理。将所得到的附着有沥青的球形化天然石墨在非活性气体中实施了1300℃热处理后,将所得到的烧成物进行破碎、分级处理,由此得到了在石墨粒子的表面含有非晶质碳物质的复合碳材料。由烧成产率确认了所得到的复合碳材料中球形化石墨粒子与非晶质碳的重量比率(球形化石墨质粒子:非晶质碳)为1:0.08。进行了与实施例a3同样的测定,将结果示于表2。
[0622]
(比较例a5)
[0623]
将平均粒径(d50)为100μm的鳞片状天然石墨粉碎,得到了d50为8.5μm的鳞片状天然石墨。在所得到的鳞片状天然石墨中混合作为造粒剂的石蜡系油12重量份后,进行球形化处理,再通过热处理除去造粒材料,得到了球形化天然石墨(平均粒径(d50)12.6μm、sa 15.3m2/g、振实密度0.88g/cm3)。将所得到的球形化石墨粉末与作为非晶质碳前体的沥青混合,在非活性气体中实施了1300℃热处理后,将烧成物进行破碎、分级处理,由此得到了负极材料,该负极材料是在石墨粒子的表面含有非晶质碳物质的复合碳材料。由烧成产率确认了所得到的负极材料(复合碳材料)中球形化石墨粒子与非晶质碳的重量比率(球形化石墨质粒子:非晶质碳)为1:0.07。进行了与实施例a3同样的测定,将结果示于表2。
[0624]
[表1]
[0625][0626]
[表2]
[0627][0628]
基于表1及表2的结果,在图1及图2中示出高温保存特性与低温输出特性的关系。在这些图表中,以实施例和比较例的结果为基础分别显示出近似直线。在各个图表中,位于
更右上的范围的结果意味着高温保存特性和低温输出特性的平衡优异。
[0629]
由表1、2及图1、2所明确的那样,具有规定的孔容、dta曲线中的热特性在规定范围内的实施例a1~a4的初始效率高、快速放电特性优异、且显示出优异的低温输出特性与高温保存特性的平衡。另一方面,对于孔容大于规定范围的比较例a1、a4、a5、孔容大于规定范围且不具有规定的热特性的比较例a2、以及不具有规定的热特性的比较例a3而言,它们的初始效率低、快速放电特性、以及低温输出特性与高温保存特性的平衡变差。另外,未进行在表面包覆非晶质碳的处理、拉曼半峰宽δνb(cm
‑1)、拉曼r2值、拉曼r3值小于规定的范围的参考例a1的初始效率低、低温输出特性与高温保存特性的平衡变差。
[0630]
[实验b]
[0631]
以下,通过实施例对本发明的内容更具体地进行说明,但本发明只要不超出其主旨,就不受以下的实施例的限定。需要说明的是,以下的实施例中的各种制造条件、评价结果的值具有作为本发明的实施方式中的上限或下限的优选的值的含义,优选的范围也可以是将前述的上限或下限的值与下述实施例的值组合、或者将实施例的值彼此组合而规定的范围。
[0632]
<负极片的制作>
[0633]
使用在后述的条件下制备的负极材料作为负极活性物质,制作了具有活性物质层密度1.5
±
0.03g/cm3的活性物质层的极板。具体地,在负极材料50.00
±
0.02g中将1重量%羧甲基纤维素钠盐水溶液50.00
±
0.02g(换算成固体成分为0.500g)、及重均分子量27万的苯乙烯
‑
丁二烯橡胶水性分散体1.00
±
0.05g(换算成固体成分为0.5g)用keyence制混合搅拌机搅拌5分钟并脱泡30秒钟,得到了浆料。
[0634]
使用模涂机将该浆料以10cm的宽度涂布在作为集电体的厚度10μm的铜箔上,使得负极材料附着10.00
±
0.2mg/cm2,干燥后,切成宽度5cm,使用直径20cm的辊进行辊压,并调整为活性物质层的密度为1.5
±
0.03g/cm3,得到了电极片。
[0635]
<正极片的制作>
[0636]
如下所述制作了正极。首先,将作为正极活性物质的镍
‑
锰
‑
钴酸锂(linimncoo2)85重量%、作为导电材料的乙炔黑10重量%、作为粘结剂的聚偏氟乙烯(pvdf)5重量%在n
‑
甲基吡咯烷酮溶剂中进行混合,得到了浆料。使用刮板涂布机将该浆料涂布在作为集电体的厚度15μm的铝箔上,使得正极材附着22.5
±
0.2mg/cm2,在130℃下进行了干燥。进一步进行辊压,调整为正极密度达到2.60
±
0.05g/cm3,得到了电极片。
[0637]
<非水二次电池(层压型电池)的制作法>
[0638]
将利用上述方法制作的附着有10.0
±
0.3mg/cm2负极材料、且活性物质层的密度为1.5
±
0.03g/cm3的负极片、正极片、及聚乙烯制隔板按照负极、隔板、正极的顺序进行了层叠。用筒状的铝层压膜包入上述得到的电池元素,注入电解液,该电解液是在碳酸亚乙酯(ec)、碳酸二甲酯(dmc)和碳酸甲乙酯(emc)的混合溶剂(体积比=3:3:4)中以1mol/l溶解有lipf6而得到的,然后进行真空密封,分别制作了片状的非水二次电池。进一步,为了提高电极间的密合性,用玻璃板夹持片状电池进行了加压。
[0639]
<非水二次电池(硬币型电池)的制作法>
[0640]
将利用上述方法制作的附着有10.0
±
0.3mg/cm2负极材料、且活性物质层的密度为1.5
±
0.03g/cm3的电极片冲裁成直径12.5mm的圆盘状,作为负极,将锂金属箔冲裁成直
径14mm的圆板状,作为对电极。在两极间放置含浸有电解液的隔板(多孔性聚乙烯膜制),分别制作了2016硬币型电池,所述电解液是在碳酸亚乙酯和碳酸甲乙酯的混合溶剂(体积比=3:7)中以1mol/l溶解有lipf6而得到的。
[0641]
<放电容量(mah/g)>
[0642]
使用利用上述方法制作的非水二次电池(2016硬币型电池),按照下面的测定方法测定了电池充放电时的容量。
[0643]
以0.05c的电流密度对锂对电极充电至5mv,再以5mv的恒定电压进行充电,直至电流密度达到0.005c,将锂掺杂到负极中后,以0.1c的电流密度对锂对电极进行了放电直至1.5v。将此时的每单位负极活性物质重量的放电容量作为该材料的放电容量(mah/g)。
[0644]
<循环膨胀>
[0645]
对于未经充放电循环的非水二次电池,在25℃以电压范围4.1v~3.0v、电流值0.2c(将用1小时将1小时率的放电容量的额定容量放完电的电流值设为1c,下同)进行3个循环,并以电压范围4.2v~3.0v、电流值0.2c(充电时以4.2v进一步实施了恒定电压充电2.5小时)进行2个循环,进行了初始充放电。在该时刻去除用于临时固定的玻璃板,利用日本三丰株式会社制造的接触式测厚仪对于1个电池测定9个点的厚度、将其平均值作为“循环前的厚度”。然后,再次用玻璃板夹入电池,然后在45℃的恒温槽中以0.8c
‑
cccv充电、0.8ccc放电、1.5v截止的条件进行25次充放电,实施了循环试验。然后,对于soc 0%的状态的电池与循环前同样的位置测定9个点,求出其平均值,作为“循环后的厚度”,并设定为:循环膨胀={(循环后的厚度)/(循环前的厚度)
‑
1}
×
100(%)。
[0646]
<快速充电特性>
[0647]
对于未经充放电循环的硬币型电池,在25℃以电压范围4.1v~3.0v、电流值0.2c(将用1小时将1小时率的放电容量的额定容量放完电的电流值设为1c,下同)进行3个循环,并以电压范围4.2v~3.0v、电流值0.2c(充电时以4.2v进一步实施了恒定电压充电2.5小时)进行2个循环,进行了初始充放电。然后,从soc 0%的状态以电流值3c进行了cc充电直至soc 100%。由得到的电压(v)
‑
容量(q)曲线的微分计算出dv/dq值,求出dv/dq值从负区域达到正区域的dv/dq=0时的容量值,设定为:快速充电特性={dv/dq=0时的容量值(mah)}/{soc 100%的容量值(mah)}
×
100(%)。
[0648]
(实施例b1)
[0649]
将平均粒径(d50)为100μm的鳞片状天然石墨粉碎,得到了d50为11.1μm的鳞片状天然石墨。相对于所得到的鳞片状天然石墨100重量份,混合了作为造粒剂的液态油12重量份,然后进行球形化处理,再通过热处理除去造粒剂,得到了球形化天然石墨。将所得到的球形化石墨填充到橡胶制容器,并进行密闭,以200mpa进行各向同性加压处理,由此控制了退汞量/进汞量(b/a)。对所得到的成型物进行了破碎、分级处理,将所得到的球形化石墨粉末(负极材料原料)的各种物性示于表1。需要说明的是,前述汞孔隙率法中的退汞量/进汞量(b/a)
×
100(%)为61.5%。将该球形化石墨粉末与作为非晶质碳前体的被调整为灰分0.02重量%、金属杂质量20重量ppm、qi 1重量%的沥青进行混合,进行减压处理使炉内压力为10torr以下,用氮进行压力恢复直至大气压,进一步流通氮而使炉内的氧浓度<100ppm,在非活性气体中实施了1300℃热处理。将所得到的烧成物进行破碎、分级处理,由此得到了在石墨粒子的表面含有非晶质碳物质的复合碳材料。由烧成产率确认了所得到的
复合碳材料中球形化石墨粒子与非晶质碳的重量比率(球形化石墨质粒子:非晶质碳)为1:0.08。将这样得到的负极材料的各种物性示于表3。另外,使用所得到的负极材料分别测定了前述的放电容量、循环膨胀、快速充电特性,将结果示于表3。需要说明的是,表3的a、b示出的是将小数第4位四舍五入而得到的值。
[0650]
(实施例b2)
[0651]
将平均粒径(d50)为100μm的鳞片状天然石墨粉碎,得到了d50为8.5μm的鳞片状天然石墨。在所得到的鳞片状天然石墨中混合作为造粒剂的液态油12重量份后,进行球形化处理,再通过热处理除去造粒剂,得到了球形化天然石墨。将所得到的球形化石墨填充到橡胶制容器,并进行密闭,以200mpa进行各向同性加压处理,由此控制了退汞量/进汞量(b/a)。对所得到的成型物进行了破碎、分级处理,将所得到的球形化石墨粉末(负极材料原料)的各种物性示于表2。需要说明的是,前述汞孔隙率法中的退汞量/进汞量(b/a)
×
100(%)为69.9%。将该球形化石墨粉末与作为非晶质碳前体的被调整为灰分0.02重量%、金属杂质量20重量ppm、qi 1重量%的沥青进行混合,进行减压处理使炉内压力为10torr以下,用氮进行压力恢复直至大气压,进一步流通氮而使炉内的氧浓度<100ppm,在非活性气体中实施了1300℃热处理。将所得到的烧成物进行破碎、分级处理,由此得到了在石墨粒子的表面含有非晶质碳物质的复合碳材料。由烧成产率确认了所得到的复合碳材料中球形化石墨粒子与非晶质碳的重量比率(球形化石墨质粒子:非晶质碳)为1:0.06。将这样得到的负极材料的各种物性示于表3。另外,使用所得到的负极材料分别测定了前述的放电容量、循环膨胀、快速充电特性,将结果示于表3。
[0652]
(比较例b1)
[0653]
将平均粒径(d50)为100μm的鳞片状天然石墨进行球形化处理,得到了球形化天然石墨(平均粒径(d50)16.2μm、bet比表面积(sa)6.9m2/g、振实密度1.00g/cm3)。所得到的球形化石墨粉末(负极材料原料)的前述汞孔隙率法中的退汞量/进汞量(b/a)
×
100(%)为27.5%。将该球形化石墨粉末与作为非晶质碳前体的被调整为灰分<0.01%、金属杂质量60ppm、qi<0.1%的焦油进行混合,进行减压处理使炉内压力为10torr以下,用氮进行压力恢复直至大气压,进一步流通氮而使炉内的氧浓度<100ppm,实施了1300℃热处理。将所得到的烧成物进行破碎、分级处理,由此得到了负极材料,该负极材料是在石墨粒子的表面含有非晶质碳物质的复合碳材料。由烧成产率确认了所得到的负极材料(复合碳材料)中球形化石墨粒子与非晶质碳的重量比率(球形化石墨质粒子:非晶质碳)为1:0.03。使用这样得到的负极材料分别测定了前述的放电容量、循环膨胀、快速充电特性,将结果示于表3。
[0654]
(比较例b2)
[0655]
将平均粒径(d50)为100μm的鳞片状天然石墨进行球形化处理,得到了球形化天然石墨(平均粒径(d50)10.9μm、bet比表面积(sa)8.1m2/g、振实密度0.90g/cm3)。所得到的球形化石墨粉末的前述汞孔隙率法中的退汞量/进汞量(b/a)
×
100(%)为28.3%。将该球形化石墨粉末与作为非晶质碳前体的被调整为灰分<0.01%、金属杂质量60ppm、qi<0.1%的焦油进行混合,进行减压处理使炉内压力为10torr以下,用氮进行压力恢复直至大气压,进一步流通氮而使炉内的氧浓度<100ppm,实施了1300℃热处理。将所得到的烧成物进行破碎、分级处理,由此得到了负极材料,该负极材料是在石墨粒子的表面含有非晶质碳物质的复合碳材料。由烧成产率确认了所得到的负极材料(复合碳材料)中球形化石墨粒子与非
晶质碳的重量比率(球形化石墨质粒子:非晶质碳)为1:0.04。使用这样得到的负极材料分别测定了前述的放电容量、循环膨胀、快速充电特性,将结果示于表3。
[0656]
(比较例b3)
[0657]
将平均粒径(d50)为100μm的鳞片状天然石墨进行球形化处理,得到了球形化天然石墨(平均粒径(d50)7.2μm、bet比表面积(sa)11.1m2/g、振实密度0.81g/cm3)。所得到的球形化石墨粉末的前述汞孔隙率法中的退汞量/进汞量(b/a)
×
100(%)为41.2%。将该球形化石墨粉末与作为非晶质碳前体的被调整为灰分<0.01%、金属杂质量60ppm、qi<0.1%的焦油进行混合,进行减压处理使炉内压力为10torr以下,用氮进行压力恢复直至大气压,进一步流通氮而使炉内的氧浓度<100ppm,实施了1300℃热处理。将所得到的烧成物进行破碎、分级处理,由此得到了负极材料,该负极材料是在石墨粒子的表面含有非晶质碳物质的复合碳材料。由烧成产率确认了所得到的负极材料(复合碳材料)中球形化石墨粒子与非晶质碳的重量比率(球形化石墨质粒子:非晶质碳)为1:0.03。使用这样得到的负极材料分别测定了前述的放电容量、循环膨胀、快速充电特性,将结果示于表3。
[0658]
(比较例b4)
[0659]
使用表面含有非晶质碳的人造石墨,分别测定了前述的放电容量、循环膨胀、快速充电特性,将结果示于表3。
[0660]
[表3]
[0661][0662]
图3示出了实施例b1~b2及比较例b1~b3的退汞量/进汞量(b/a)与循环膨胀的关系。基于表3、图3可知,通过使用本发明的第2实施方式的负极材料原料及负极材料,可以得到放电容量高、且循环膨胀的抑制、快速充电特性优异的锂离子二次电池。
[0663]
[实验c]
[0664]
以下,通过实施例对本发明的内容更具体地进行说明,但本发明只要不超出其主旨,就不受以下的实施例的限定。需要说明的是,以下的实施例中的各种制造条件、评价结果的值具有作为本发明的实施方式中的上限或下限的优选的值的含义,优选的范围也可以是将前述的上限或下限的值与下述实施例的值组合、或者将实施例的值彼此组合而规定的范围。
[0665]
<负极片的制作>
[0666]
使用在后述的条件下制备的负极材料作为负极活性物质,制作了具有活性物质层密度1.65
±
0.03g/cm3的活性物质层的极板。具体地,在负极材料50.00
±
0.02g中将1重量%羧甲基纤维素钠盐水溶液50.00
±
0.02g(换算成固体成分为0.500g)、及重均分子量27万的苯乙烯
‑
丁二烯橡胶水性分散体1.00
±
0.05g(换算成固体成分为0.5g)用keyence制混合搅拌机搅拌5分钟并脱泡30秒钟,得到了浆料。
[0667]
使用模涂机将该浆料以10cm的宽度涂布在作为集电体的厚度10μm的铜箔上,使得
负极材料附着10.00
±
0.2mg/cm2,干燥后,切成宽度5cm,使用直径20cm的辊进行辊压,并调整为活性物质层的密度为1.5
±
0.03g/cm3及1.65
±
0.03g/cm3,得到了各自的电极片。将制作活性物质层的密度为1.65g/cm3的电极片时施加的载荷的峰值设为加压载荷[kgf/5cm],示于表4中。
[0668]
<正极片的制作>
[0669]
如下所述制作了正极。首先,将作为正极活性物质的镍
‑
锰
‑
钴酸锂(linimncoo2)85重量%、作为导电材料的乙炔黑10重量%、作为粘结剂的聚偏氟乙烯(pvdf)5重量%在n
‑
甲基吡咯烷酮溶剂中进行混合,得到了浆料。使用刮板涂布机将该浆料涂布在作为集电体的厚度15μm的铝箔上,使得正极材附着22.5
±
0.2mg/cm2,在130℃下进行了干燥。进一步进行辊压,调整为正极密度达到2.60
±
0.05g/cm3,得到了电极片。
[0670]
<非水二次电池(层压型电池)的制作法>
[0671]
将利用上述方法制作的附着有10.0
±
0.3mg/cm2负极材料、且活性物质层的密度为1.65
±
0.03g/cm3(的负极片和正极片)、以及聚乙烯制隔板按照负极、隔板、正极的顺序进行了层叠。用筒状的铝层压膜包入上述得到的电池元素,注入电解液,该电解液是在碳酸亚乙酯(ec)、碳酸二甲酯(dmc)和碳酸甲乙酯(emc)的混合溶剂(体积比=3:3:4)中以1mol/l溶解有lipf6而得到的,然后进行真空密封,分别制作了片状的非水二次电池。进一步,为了提高电极间的密合性,用玻璃板夹持片状电池进行了加压。
[0672]
<膨胀测定>
[0673]
对于未经充放电循环的非水二次电池,在25℃以电压范围4.1v~3.0v、电流值0.2c(将用1小时将1小时率的放电容量的额定容量放完电的电流值设为1c,下同)进行3个循环,并以电压范围4.2v~3.0v、电流值0.2c(充电时以4.2v进一步实施了恒定电压充电2.5小时)进行2个循环,进行了初始充放电。在该时刻去除用于临时固定的玻璃板,利用日本三丰株式会社制造的接触式测厚仪对于1个电池测定9个点的厚度、将其平均值作为循环前的厚度。然后,再次用玻璃板夹入电池,然后在45℃的恒温槽中以0.8c
‑
cccv充电、0.8ccc放电、1.5v截止的条件进行25次充放电,实施了循环试验。然后,对于soc 0%的状态的电池与循环前同样的位置测定9个点,求出其平均值,将其与循环前的厚度之差作为循环中的极板的膨胀,将其差值示于表4。
[0674]
(实施例c1)
[0675]
将平均粒径(d50)为100μm的鳞片状天然石墨粉碎,得到了d50为9μm的鳞片状天然石墨。在所得到的鳞片状天然石墨中混合作为造粒剂的液态油12重量份后,进行球形化处理,再通过热处理除去造粒材料,得到了球形化天然石墨(平均粒径(d50)13μm、sa19m2/g、振实密度0.94g/cm3)。将所得到的球形化天然石墨填充到橡胶制容器,并进行密闭,以200mpa进行各向同性加压处理后,对所得到的成型物进行了破碎、分级处理。将所得到的球形化天然石墨粉末与作为非晶质碳前体的被调整为灰分0.02重量%、金属杂质量20重量ppm、qi 1重量%的沥青进行混合,进行减压处理使炉内压力为10torr以下,用氮进行压力恢复直至大气压,进一步流通氮而使炉内的氧浓度<100ppm,在非活性气体中实施了1300℃热处理。将所得到的烧成物进行破碎、分级处理,由此得到了负极材料,该负极材料是在石墨粒子的表面含有非晶质碳物质的复合碳材料。由烧成产率确认了所得到的负极材料(复合碳材料)中球形化石墨粒子与非晶质碳质物质的重量比率(球形化石墨质粒子:非晶
质碳质物质)为1:0.06。另外,对于所得到的负极材料,按照上述的方法测定了振实密度、压片密度、真密度、微孔分布,将结果示于表4。表4中,“峰顶微孔径”是微孔径小的一侧的峰的峰顶的微孔径。另外,将压汞法测定结果示于图6。
[0676]
(实施例c2)
[0677]
将平均粒径(d50)为100μm的鳞片状天然石墨粉碎,得到了d50为11μm的鳞片状天然石墨。在所得到的鳞片状天然石墨中混合作为造粒剂的液态油12重量份后,进行球形化处理,再通过热处理除去造粒材料,得到了球形化天然石墨(平均粒径(d50)16μm、sa 15m2/g、振实密度0.96g/cm3)。将所得到的球形化石墨填充到橡胶制容器,并进行密闭,以200mpa进行各向同性加压处理后,对所得到的成型物进行了破碎、分级处理。将所得到的球形化石墨粉末与作为非晶质碳前体的被调整为灰分0.02重量%、金属杂质量20重量ppm、qi 1重量%的沥青进行混合,进行减压处理使炉内压力为10torr以下,用氮进行压力恢复直至大气压,进一步流通氮而使炉内的氧浓度<100ppm,在非活性气体中实施了1300℃热处理。将所得到的烧成物进行破碎、分级处理,由此得到了负极材料,该负极材料是在石墨粒子的表面含有非晶质碳物质的复合碳材料。由烧成产率确认了所得到的负极材料(复合碳材料)中球形化石墨粒子与非晶质碳质物质的重量比率(球形化石墨质粒子:非晶质碳质物质)为1:0.06。对于所得到的负极材料,进行了与实施例c1同样的测定,将结果示于表4。
[0678]
(实施例c3)
[0679]
除了使球形化石墨粒子与非晶质碳的重量比率(球形化石墨质粒子:非晶质碳物质)为1:0.08以外,按照与实施例c2同样的方法得到了负极材料(复合碳材料)。对于所得到的负极材料,进行了与实施例c1同样的测定,将结果示于表4。
[0680]
(比较例c1)
[0681]
将对平均粒径(d50)为100μm的鳞片状天然石墨进行球形化处理而制作的球形化天然石墨(平均粒径(d50)16μm、bet比表面积(sa)6.9m2/g、振实密度1.0g/cm3)填充于橡胶制容器,并进行密闭,以100mpa进行各向同性加压处理后,对所得到的成型物进行了破碎、分级处理。将所得到的球形化天然石墨粉末与作为非晶质碳前体的被调整为灰分<0.01%、金属杂质量60ppm、qi<0.1%的焦油进行混合,进行减压处理使炉内压力为10torr以下,用氮进行压力恢复直至大气压,进一步流通氮而使炉内的氧浓度<100ppm,实施了1300℃热处理。将所得到的烧成物进行破碎、分级处理,由此得到了负极材料,该负极材料是在石墨粒子的表面含有非晶质碳物质的复合碳材料。由烧成产率确认了所得到的负极材料(复合碳材料)中球形化石墨粒子与非晶质碳质物质的重量比率(球形化石墨质粒子:非晶质碳质物质)为1:0.03。对于所得到的负极材料,进行了与实施例c1同样的测定,将结果示于表4。另外,将压汞法测定结果示于图6。
[0682]
(比较例c2)
[0683]
除了未进行各向同性加压处理以外,按照与实施例c1同样的方法得到了作为复合碳材料的负极材料。对于所得到的负极材料(复合碳材料),进行了与实施例c1同样的测定,将结果示于表4。
[0684]
(比较例c3)
[0685]
未进行各向同性加压处理,并且球形化石墨粒子与非晶质碳物质的重量比率(球形化石墨质粒子:非晶质碳物质)为1:0.04,除此以外,按照与比较例同样的方法得到了作
为复合碳材料的负极材料。对于所得到的负极材料(复合碳材料),进行了与实施例c1同样的测定,将结果示于表4。
[0686]
(比较例c4)
[0687]
使用球形化天然石墨(平均粒径(d50)11μm、bet比表面积(sa)8.2m2/g、振实密度0.88g/cm3),未进行各向同性加压处理,且使用调整为qi=2.5%的煤焦油作为非晶质碳前体,球形化石墨粒子与非晶质碳物质的重量比率(球形化石墨质粒子:非晶质碳物质)为1:0.07,除此以外,按照与实施例c4同样的方法得到了负极材料。对于所得到的负极材料,进行了与实施例c1同样的测定,将结果示于表4。
[0688]
(比较例c5)
[0689]
将对平均粒径(d50)为100μm的鳞片状天然石墨进行球形化处理而制作的球形化天然石墨(平均粒径(d50)13μm、bet比表面积(sa)7.8m2/g、振实密度0.93g/cm3)与作为石墨质物质前体的调整为灰分0.1重量%、qi<0.2重量%的沥青进行混合后,填充于橡胶制容器并进行密闭,然后进行了各向同性加压处理。然后,使氮流通并在1000℃进行热处理而脱焦油,然后在非活性气氛下于3000℃实施了石墨化处理。对所得到的石墨化物进行破碎、分级处理,得到了负极材料,该负极材料是在石墨粒子的表面或内部含有石墨质物质的复合碳材料。由烧成产率确认了所得到的作为复合碳材料的负极材料中球形化石墨粒子与石墨质物质的重量比率(球形化石墨质粒子:石墨质物质)为1:0.10。对于所得到的负极材料(复合碳材料),进行了与实施例c1同样的测定,将结果示于表4。
[0690]
(比较例c6)
[0691]
将针状生焦粉碎成10μm,使氮流通在1000℃进行热处理而脱焦油后,在非活性气氛下于3000℃实施石墨化处理,得到了针状的人造石墨(平均粒径(d50)11μm、bet比表面积(sa)1.8m2/g、振实密度1.1g/cm3)。将所得到的人造石墨与作为非晶质碳前体的调整为灰分<0.01%、金属杂质量60ppm、qi<0.1%的焦油混合后,进行减压处理使炉内压力为10torr以下,用氮进行压力恢复直至大气压,进一步流通氮而使炉内的氧浓度<100ppm,实施了1300℃热处理。由烧成产率确认了所得到的作为复合碳材料的负极材料中球形化石墨粒子与石墨质物质的重量比率(球形化石墨质粒子:石墨质物质)为1:0.04。对于所得到的负极材料(复合碳材料),进行了与实施例c1同样的测定,将结果示于表4。
[0692]
[表4]
[0693][0694]
图4中横轴设为覆盖率、纵轴设为压汞法的极小值以下的孔容,示出了覆盖率与孔容得关系。在图4中还一并示出了式(1)及式(2)。图5中,横轴设为(压片密度)
‑
(振实密度)
(图4中,以“pellet
‑
tap”表示)、纵轴设为实际的“加压载荷”(压制成1.65g/cm3时所必须的加压载荷),示出了pellet
‑
tap与加压载荷的关系。由图5可知,满足特定的式(1)、式(2)的材料如图5所示那样压制性良好。
[0695]
由表4、及图4、5所明确的那样,在为包含在表面的至少一部分具有非晶质碳物质或石墨质物质中的至少一者的石墨的负极材料、且由压汞法测得的累积孔容与石墨表面的非晶质碳物质及石墨质物质的覆盖率满足特定式的实施例的情况下,确认到良好的压制性、且循环时极板的膨胀收缩非常小。另一方面,确认到比较例c1的压制性虽然良好,但极板膨胀大。确认到比较例c2虽然极板膨胀小,但由于为式(1)、式(2)的范围外,压制载荷大。
[0696]
即,可知通过满足以下两者,即满足式(1)、式(2)、且微孔径小的一侧的峰的峰顶的微孔径为特定范围内,能够压制加工成高密度,且表现出循环时的极板膨胀小这样的性能。
[0697]
工业实用性
[0698]
本发明的第1实施方式的非水二次电池用负极材料、以及包含其的非水二次电池用负极及非水二次电池由于是高容量的,且快速充放电特性、低温输入输出特性、高温保存特性优异,因此可以适用于车载用途;电动工具用途;手机、个人电脑等便携机器用途等。
[0699]
本发明的第2实施方式的非水二次电池用负极材料原料及非水二次电池用负极材料、以及包含其的非水二次电池用负极及非水二次电池由于是高容量的、且循环膨胀小、快速充放电特性优异,因此可以适用于车载用途;电动工具用途;手机、个人电脑等便携机器用途等。
[0700]
本发明的第3实施方式的非水二次电池用负极材料、以及包含其的非水二次电池用负极及非水二次电池作为面向高容量的电池用负极材料,其快速充放电特性、低温输入输出特性、高温保存特性优异,因此可以适用于车载用途;电动工具用途;手机、个人电脑等便携机器用途等。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1