Co-Mo双金属氮化物氧还原催化剂及其制备方法和应用
本发明涉及一种co-mo双金属氮化物氧还原催化剂及其制备方法和应用,属于燃料电池阴极氧还原催化剂领域。
背景技术:
质子交换膜燃料电池(pemfc)作为一种再生能源设备,可将燃料和氧化剂的化学能直接转换为电能,具有高效、高可靠性、低排放、静音运行等特点。氧还原反应(orr)作为燃料电池中最关键的反应之一,仍然受到许多挑战的困扰,例如反应动力学缓慢和电化学不稳定等。当前,贵金属特别是基于pt的催化剂被认为是orr最先进的电催化剂。然而,其小分子抗毒性和稳定性差,以及这些贵金属的高成本阻碍了它们的大规模商业应用。
过渡金属氮化物(tmns)是一种新的非贵金属基催化剂,由于其表面和吸附性能与pt基催化剂相似,因此被视为“类铂”催化剂。此外,由于tmns具有良好的导电性、耐蚀性、高熔点和优异的热稳定性等固有优势,可用于制备有潜力的电催化剂。然而,orr性能与pt/c催化剂相比仍然有很大差异。通过引入另一种具有良好电负性和氧吸附能力的过渡金属以形成双金属氮化物(btmns),在金属氮化物和其他金属之间建立协同效应,可以增强基于金属氮化物材料的氧还原性能。目前,主要在前驱体制备时加入含氮试剂,如atthaponsrifa等人[thejournalofphysicalchemistryb,2012,117,(2):858-865]在制备co3mo3n催化剂过程中添加含六亚甲基四胺,而该试剂易燃,且易腐蚀皮肤,不利于安全制备。此外,zhang等人[thejournalofphysicalchemistryb,2012,117(2):858-865]在制备co3mo3n作为锂氧气电池催化剂时,氮化时的升温程序复杂(室温至350℃,5℃min-1;350至450℃,0.5℃min-1;450至800℃,2℃min-1),不具有普适性。另外,中国发明专利[cn201910792031.8]采用热分解法制备ni2w3n双金属氮化物,该氮化环境是在氮气和氢气混合气体中煅烧,而氢气是一种在高温条件下极易爆炸的危险气氛。综上所述,上述方法均不利于工业化。
技术实现要素:
本发明的目的在于提供一种低成本、高活性和高稳定性的co-mo双金属氮化物氧还原催化剂及其制备方法,以及其在氧还原催化反应中的应用。
实现本发明目的的技术方案如下:
co-mo双金属氮化物氧还原催化剂的制备方法,先通过水热法制备comoo4前驱体粉末,其形貌通过尿素的添加量控制,然后在nh3气流下氮化,得到co3mo3n氧还原催化剂,具体步骤如下:
步骤(1),按等摩尔金属比例,将钴盐、钼盐溶解于水中,室温下搅拌均匀,形成混合过渡金属盐溶液;
步骤(2),将混合过渡金属盐溶液在160±5℃下进行水热反应,反应结束后,降至室温,依次用水和乙醇离心洗涤,干燥,得到双金属氧化物comoo4的前驱体固体粉末;
步骤(3),将双金属氧化物comoo4的前驱体固体粉末在600~1000℃的nh3气氛下进行氮化处理,冷却后得到双金属氮化物co3mo3n催化剂。
步骤(1)中,钴盐为氯化钴、硝酸钴或醋酸钴;钼盐为钼酸铵或钼酸钠。
步骤(1)中,混合过渡金属盐溶液中还添加尿素。优选地,尿素与金属钴的摩尔比为1~5:1。
步骤(2)中,水热反应时间为24±2h。
步骤(2)中,干燥温度为200±10℃。
步骤(3)中,氮化处理时间为2±0.5h。
优选地,步骤(3)中,氮化温度为700℃。
本发明还提供上述制备方法制得的co-mo双金属氮化物氧还原催化剂。
进一步地,本发明提供上述co-mo双金属氮化物氧还原催化剂在氧还原催化反应中的应用。
与现有技术相比,本发明具有以下优点:
(1)采用简单的两步法,按等摩尔金属比例配制金属盐溶液,水热得到双金属氧化物,通过尿素调节形貌,然后通过nh3氮化得到双金属氮化物,制备方法安全简单,产率高,并且产物为纯相co3mo3n,物相较纯。
(2)本发明制备的co-mo双金属氮化物氧还原催化剂在碱性环境中表现出优异的氧还原催化性能,并且有可观的耐甲醇性能和稳定性。具体地,co-mo双金属氮化物材料在催化orr反应时,起始电位达到0.85vvs.rhe,与商业pt/c接近。此外其表现出比商业pt/c更为优异的稳定性,在碱性环境0.7v电压下进行12000s的计时电流耐久性测试的结果显示,其电流密度仅损失9.4%,而商业pt/c的电流密度降低了近22.4%。
附图说明
图1是干燥处理得到的comoo4的xrd图;
图2是双金属氮化物co3mo3n催化剂的xrd图;
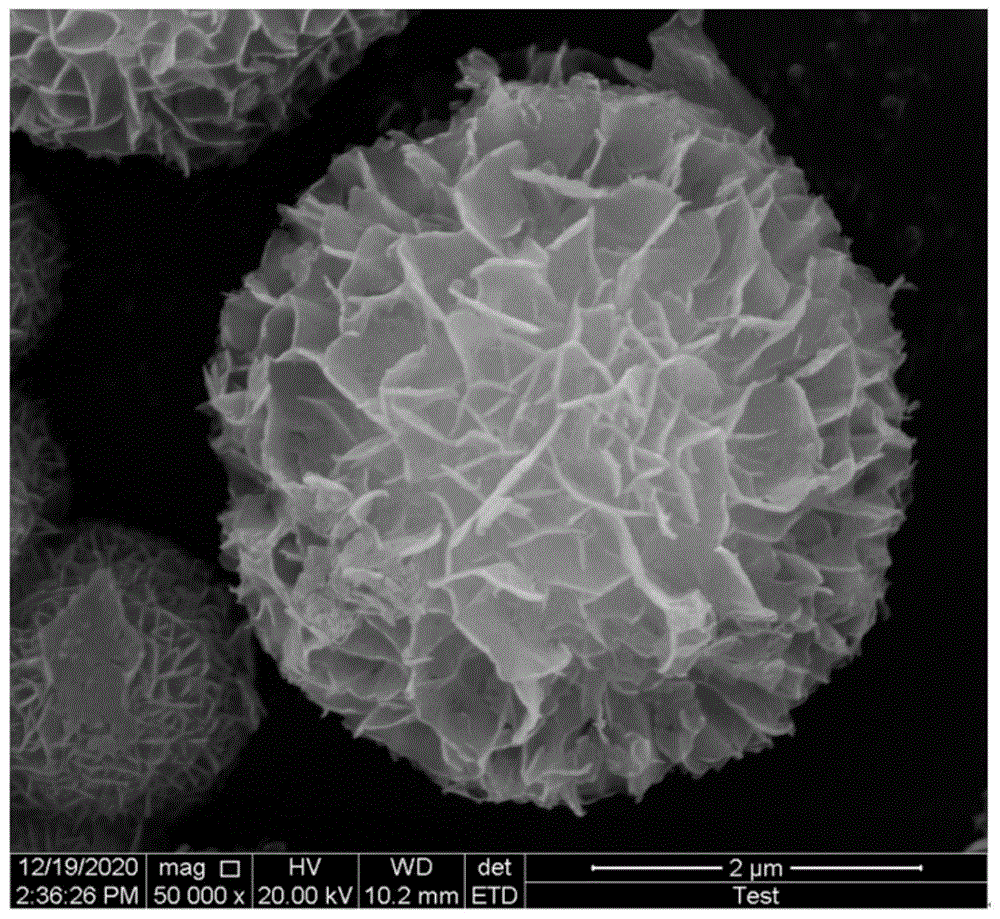
图3是实施例1得到的comoo4的sem图;
图4是实施例2得到的comoo4的sem图;
图5是实施例3得到的comoo4的sem图;
图6是实施例1、2、3不同尿素添加量制备的催化剂以及商业pt/c的线性扫描伏安(lsv)图;
图7是实施例1、4、5和对比例1、2不同氮化温度制备的催化剂的lsv图;
图8是实施例1和对比例3、4制备的催化剂的lsv图;
图9是实施例1和商业pt/c的稳定性测试结果图;
图10是实施例1和商业pt/c的耐甲醇测试结果图。
具体实施方式
下面结合具体实施例和附图对本发明作进一步说明。
实施例1
第一步,称取0.6mmol的cocl2·6h2o和0.6mmol(指的是金属mo离子的浓度,下同)的h24mo7n6o24·4h2o溶解于去离子水中,再加入3mmol的尿素,在室温下搅拌均匀。
第二步,将第一步的混合溶液在160±5℃下进行水热反应,反应结束后,降至室温,依次用水和乙醇离心洗涤,200℃下干燥,得到双金属氧化物comoo4的前驱体固体粉末;
第三步,将双金属氧化物comoo4的前驱体固体粉末置于磁舟中,在700℃的nh3气氛下氮化处理2±0.5h,随后自然降温至室温,得到黑色氮化物催化材料co3mo3n。
对本实施例1中得到的comoo4和co3mo3n进行表征测试,其xrd测试结果如图1和图2所示,制得纯相前驱体和催化剂,无杂质相。图3的sem图显示出加入过量尿素得到的comoo4呈花瓣微球,由均匀的纳米片组成。
实施例2
第一步,称取0.6mmol的cocl2·6h2o和0.6mmol的h24mo7n6o24·4h2o溶解于去离子水中,再加入0.6mmol的尿素,在室温下搅拌均匀。
第二步,将第一步的混合溶液在160±5℃下进行水热反应,反应结束后,降至室温,依次用水和乙醇离心洗涤,200℃下干燥,得到双金属氧化物comoo4的前驱体固体粉末;
第三步,将双金属氧化物comoo4的前驱体固体粉末置于磁舟中,在700℃的nh3气氛下氮化处理2±0.5h,随后自然降温至室温,得到黑色氮化物催化材料co3mo3n。
对本实施例3中干燥处理得到的comoo4进行sem测试,如图4所示,在加入与金属离子相同浓度的尿素时,comoo4由大小不匀的薄片组成。
实施例3
第一步,称取0.6mmol的cocl2·6h2o和0.6mmol的h24mo7n6o24·4h2o溶解于去离子水中,在室温下搅拌均匀。
第二步,将混合溶液在160±5℃下进行水热反应,反应结束后,降至室温,依次用水和乙醇离心洗涤,200℃下干燥,得到双金属氧化物comoo4的前驱体固体粉末;
第三步,将双金属氧化物comoo4的前驱体固体粉末置于磁舟中,在700℃的nh3气氛下氮化处理2±0.5h,随后自然降温至室温,得到黑色氮化物催化材料co3mo3n。
对本实施例3中干燥处理得到的comoo4进行sem测试,如图5所示,在不加入尿素时,为细长的棒状形貌。
实施例4
本实施例与实施例1基本相同,唯一不同的是氮化温度是600℃。
实施例5
本实施例与实施例1基本相同,唯一不同的是氮化温度是1000℃。
对比例1
本对比例与实施例1基本相同,唯一不同的是氮化温度是500℃
对比例2
本对比例与实施例1基本相同,唯一不同的是氮化温度是1100℃。
对比例3
第一步,称取0.6mmol的cocl2·6h2o和2.4mmol的h24mo7n6o24·4h2o溶解于去离子水中,再加入0.6mmol的尿素,在室温下搅拌均匀。
第二步,将第一步的混合溶液在160±5℃下进行水热反应,反应结束后,降至室温,分别用水和乙醇离心洗涤,冷冻干燥,得到前驱体固体粉末;
第三步,将前驱体固体粉末置于磁舟中,在700℃的nh3气氛下氮化处理2±0.5h,随后自然降温至室温,得到相应氮化物催化剂。
对比例4
第一步,将称取的0.6mmol的cocl2·6h2o溶解于去离子水中,在室温下搅拌均匀。
第二步,将上述溶液在160±5℃下进行水热反应,反应结束后,降至室温,分别用水和乙醇离心洗涤,冷冻干燥,得到前驱体固体粉末;
第三步,将前驱体固体粉末置于磁舟中,在700℃的nh3气氛下氮化处理2±0.5h,随后自然降温至室温,得到相应氮化物催化剂。
应用例
对实施例及对比例制备得到的催化剂样品以及商业pt/c催化剂进行氧还原性能测试,具体方法如下:
5.0mg制备的催化剂材料加入1.0ml0.05wt%nafion乙醇溶液中,室温超声1h使催化剂均匀分散在溶液中,呈墨水状。然后将30.0μl制备的催化剂溶液分次均匀滴加在直径为5.0mm的旋转圆盘电极(rde)表面,自然干燥后得到工作电极。电化学测试在室温三电极体系下进行,以制得的rde为工作电极、碳棒为对电极、饱和甘汞电极(sce)为参比电极,选择0.1mol/l的koh作为电解液。
如图6所示,对实施例1、2、3不同尿素添加量制备的催化剂以及商业pt/c进行lsv测试,很明显发现,前驱体为花状得到的co3mo3n的起始电位(0.85vvs.rhe,下同)>前驱体为片状得到的co3mo3n的起始电位(0.80v)>前驱体为棒状得到的co3mo3n的起始电位(0.76v),说明尿素的添加量调控前驱体形貌对氧还原性能的作用很大。
如图7所示,对实施例1、4、5和对比例1、2不同氮化温度制备进行催化剂的lsv,可以看出,在600-1000℃的氮化温度范围内,其氧还原的半波电位较该范围外(大于1000℃或低于600℃)的更正。具体地,700℃样品(0.75v)>600℃样品(0.71v)>1000℃样品(0.69v)>>1100℃样品(0.61v)≈500℃样品(0.60v),说明其氮化温度对氧还原性能的影响非常大。另外,在600-1000℃的氮化范围内,700℃时的半波电位更正,说明在此氮化范围内,700℃氮化温度得到的催化剂的氧还原性能最佳。
如图8所示,对实施例1和对比例3、4制备的催化剂进行lsv测试,将实施例1与对比例3的性能对比发现,对比例3的起始电位为0.75v,不如最佳实施例1(0.85v)好,说明等摩尔比例金属盐投料时得到的双金属氮化物的性能更好。此外,实施例1和对比例4比较发现,对比例4的单金属氮化物氧还原性能(0.72v)也远不如实施例1(0.85v)得到的双金属氮化物的性能好,这是由于引入第二相金属的协同效应对氧还原性能有促进作用。
如图9所示,利用计时电流法对实施例1得到的co3mo3n和商业pt/c催化剂进行稳定性能测试。在经过12000s的计时电流测试后,co3mo3n电流密度保持在90.6%,而pt/c电流保持率仅有77.6%,说明本发明得到的co3mo3n有优异的稳定性。
如图10所示,利用计时电流法对实施例1得到的co3mo3n和商业pt/c催化剂进行耐甲醇性能测试。co3mo3n在甲醇加入后基本稳定,电流密度基本保持不变;相反,pt/c受到甲醇的严重毒化作用,电流密度急剧变化,说明本发明得到的co3mo3n具有优异的耐甲醇作用。
综上所述,通过相同金属离子浓度的金属盐可顺利制备纯相的comoo4前驱体,通过调节尿素的添加量控制形貌。氮化后制备的纯相co3mo3n具有优异的氧还原性能,耐甲醇毒化能力及稳定性。
- 还没有人留言评论。精彩留言会获得点赞!