软磁性合金和磁性部件的制作方法
1.本发明涉及软磁性合金、以及使用了该软磁性合金的磁性部件。
背景技术:
2.作为电感器等各种磁性部件中使用的磁性材料,已知专利文献1~3所示的软磁性合金。这些软磁性合金的饱和磁通密度bs比铁氧体材料高,并且具有良好的软磁特性。但是,软磁性合金有时由于保存状态或使用环境而发生生锈等腐蚀,需要提高耐腐蚀性。
3.现有技术文献
4.专利文献
5.专利文献1:日本特开2009-293099号公报
6.专利文献2:日本特开2007-231415号公报
7.专利文献3:日本特开2014-167139号公报
技术实现要素:
8.发明所要解决的技术问题
9.本发明是鉴于上述情况而完成的,其目的在于提供一种具有高耐腐蚀性的软磁性合金、以及使用了该软磁性合金的磁性部件。
10.用于解决技术问题的技术方案
11.为了实现上述目的,本发明提供一种软磁性合金,其具有:
12.内部区域,其具有含有fe和p(磷)的软磁性的合金组成;和
13.p浓化区域,其存在于比上述内部区域靠表面侧,p的浓度比上述内部区域高。
14.本发明的发明人进行了深入研究,结果发现利用具有上述特征的软磁性合金,能够抑制浸水时生锈,耐腐蚀性提高。
15.优选上述p浓化区域含有与上述内部区域共同的元素,
16.上述p浓化区域中的与上述内部区域共同的元素的合计含量以物质的量比换算计为50%以上。
17.优选上述内部区域含有co,co的浓化区域存在于比上述内部区域靠表面侧,上述co的浓化区域与上述p浓化区域至少部分重合。而且,优选上述co的浓化区域为金属相,优选上述co的浓化区域中的co浓化度大于1.2。
18.另外,优选上述p浓化区域中的p浓化度为1.5以上,更优选为2.0以上。
19.另外,优选上述软磁性合金的非晶质化度为85%以上。
20.上述软磁性合金可以具有薄带形状,也可以具有粉末形状。
21.本发明的软磁性合金的用途没有特别限制,例如,能够适用于电感器等的线圈部件、滤波器、天线等的各种磁性部件。本发明的软磁性合金也适合在上述用途中作为线圈部件等的磁芯(magnetic core)材料。
附图说明
22.图1a是将本发明的一个实施方式的软磁性合金1的主要部分放大的截面图。
23.图1b是将本发明的一个实施方式的软磁性合金1a的主要部分放大的截面图的一例。
24.图2a是通过x射线结晶结构解析得到的图的一例。
25.图2b是对图2a所示的图进行图形拟合而得到的图案的一例。
26.图3a是沿着图1a所示的测定线lm,使用edx进行线分析而得到的曲线的一例。
27.图3b是沿着图1b所示的测定线lma,使用edx进行线分析而得到的曲线的一例。
28.图4a是表示本发明的一个实施方式的软磁性合金1b的截面图。
29.图4b是将图4a所示的区域ivb放大的截面图。
30.图5a是图1a所示的软磁性合金1的eels图像的一例。
31.图5b是图1b所示的软磁性合金1a的eels图像的一例。
32.图5c是图4a所示的软磁性合金1b的stem图像的一例。
33.附图标记说明
34.1、1a、1b:软磁性合金;2:内部区域;10:最表面;11:浓化部;11a:p浓化区域;11b:co浓化区域;12:sb氧化层;13:fe氧化层;20:覆盖层。
具体实施方式
35.以下,基于附图所示的实施方式详细地说明本发明。
36.本实施方式的软磁性合金1可以具有薄带形状、粉末形状、或其它的块体形状等,软磁性合金1的形状没有特别限定。另外,软磁性合金1的尺寸也没有特别限定。例如,在软磁性合金1为薄带形状的情况下,薄带的厚度可以为15μm~100μm;在软磁性合金1为粉末形状的情况下,该软磁性合金粉末的平均粒径可以为0.5μm~150μm,优选为0.5μm~25μm。
37.其中,上述的平均粒径可以通过激光衍射法等各种粒度分析法进行测定,优选使用颗粒图像分析装置morphologi g3(malvern panalytical公司制造)进行测定。使用morphologi g3,利用空气使软磁性合金粉末分散,测定构成该粉末的颗粒的投影面积,根据该投影面积得到圆当量直径的粒度分布。然后,在所得到的粒度分布中,计算体积基准或个数基准的累积相对度数达到50%的粒径作为平均粒径即可。其中,在软磁性合金1包含于磁芯中的情况下,软磁性合金1(粉末)的平均粒径可以通过使用电子显微镜(sem、stem等)的截面观察测量截面所包括的颗粒的圆当量直径算出。
38.图1a是将软磁性合金1的表面附近放大的截面图。如图1a所示,软磁性合金1具有内部区域2、和位于比该内部区域2靠软磁性合金1的表面侧的浓化部11。其中,在本实施方式中,“内侧”是指更接近软磁性合金1的中心的一侧,“表面侧”或“外侧”是指更远离软磁性合金1的中心的一侧。
39.(内部区域2)
40.内部区域2是占软磁性合金1的体积中的至少90vol%以上的软磁性合金1的基体部。因此,软磁性合金1的平均组成可以视为内部区域2的组成,软磁性合金1的结晶结构可以视为内部区域2的结晶结构。此外,上述的内部区域2的体积比例能够代替面积比例,软磁性合金1的截面面积中的至少90%以上为内部区域2。
41.内部区域2(即软磁性合金1)具有含有fe和p(磷)的软磁性的合金组成,内部区域2中的p的含有率优选为0.1at%~10at%,更优选为2.0at%~7.0at%。另外,优选在内部区域2中除了fe和p以外还含有co。
42.内部区域2的具体的合金种类没有特别限定,例如可以为fe-co系合金或fe-co-v系合金、fe-co-si系合金、fe-co-si-al系合金、fe-co-si-cr系合金等含p的结晶系的软磁性合金。另外,从降低矫顽力的观点来看,内部区域2优选具有非晶质或纳米结晶的合金组成,作为非晶质或纳米结晶的软磁性合金,可以举出fe-co-p-c系合金、fe-co-b-p系合金或fe-co-b-si-p系合金等。进一步具体而言,内部区域2更优选具有满足组成式((fe
(1-(α+β)
co
α
ni
β
)
1-γ
x1
γ
)
(1-(a+b+c+d+e))
bapbsiccdcre的合金组成,通过具有上述组成,容易得到非晶质、异质非晶(heteroamorphous)或纳米结晶的结晶结构。
43.在上述组成式中,b为硼,p为磷,c为碳,x1为选自ti、zr、hf、nb、ta、mo、w、al、ga、ag、zn、s、ca、mg、v、sn、as、sb、bi、n、o、au、cu、稀土元素和铂族元素中的1种以上的元素。稀土元素包括sc、y和镧系元素,铂族元素包括ru、rh、pd、os、ir和pt。另外,α、β、γ、a、b、c、d、e为原子数比,这些原子数比优选满足以下条件。
44.co相对于fe的含量(α)为0≤α≤0.700,可以为0.005≤α≤0.600,也可以为0.030≤α≤0.600,还可以为0.050≤α≤0.600。通过α在上述范围内,bs和耐腐蚀性提高。从提高bs的观点来看,优选为0.050≤α≤0.500。虽然存在α越大耐腐蚀性越高的倾向,但在α过大的情况下,bs容易降低。
45.另外,ni相对于fe的含量(β)为0≤β≤0.200。即,可以不含ni,也可以为0.005≤β≤0.200。从提高bs的观点来看,可以为0≤β≤0.050,也可以为0.001≤β≤0.050,还可以为0.005≤β≤0.010。虽然存在β越大耐腐蚀性越高的倾向,但在β过大的情况下,bs降低。
46.x1可以作为杂质含有,也可以有意地添加。x1的含量(γ)为0≤γ<0.030。即,相对于fe、co和ni的合计含量,可以利用x1置换低于3.0%。
47.另外,将构成软磁性合金的各元素的原子数比之和设为1时,fe、co、ni和x1的合计含量的原子数比(1-(a+b+c+d+e))优选为0.720≤(1-(a+b+c+d+e))≤0.950,更优选为0.780≤(1-(a+b+c+d+e))≤0.890。通过满足该条件,bs容易提高。另外,通过为0.720≤(1-(a+b+c+d+e))≤0.890,容易得到非晶质。
48.a为b的原子数比,优选为0≤a≤0.200,从提高bs的观点来看,更优选为0≤a≤0.150。
49.b为p的原子数比,优选为0.001≤b≤0.100,从同时提高bs和耐腐蚀性的观点来看,更优选为0.005≤b≤0.080,进一步优选为0.005≤b≤0.050。
50.c为si的原子数比,优选为0≤c≤0.150。即,可以不含si,从同时提高bs和耐腐蚀性的观点来看,更优选为0.001≤c≤0.070。
51.d为c的原子数比,优选为0≤d≤0.050。即,可以不含c,从同时提高bs和耐腐蚀性的观点来看,更优选为0≤d≤0.020。
52.e为cr的原子数比,优选为0≤e≤0.050。即,从提高bs的观点来看,可以不含cr,从同时提高bs和耐腐蚀性的观点来看,更优选为0.001≤e≤0.020。
53.上述的内部区域2的组成(即软磁性合金1的组成)可以使用例如电感耦合等离子体发射光谱分析(icp)进行分析。此时,在难以利用icp求取氧量的情况下,可以并用脉冲加
热熔融提取法。另外,在难以利用icp求取碳量和硫量的情况下,可以并用红外吸收法。
54.另外,除了icp之外,也可以利用电子显微镜所附带的edx(能量色散x射线分析)或epma(电子探针显微分析仪)实施组成分析。例如,对于具有树脂成分的磁芯中所包含的软磁性合金1,有时难以利用icp进行组成分析,在该情况下,可以使用edx或epma进行组成分析。另外,在利用上述任一种方法均难以进行详细的组成分析的情况下,也可以使用3dap(三维原子探针)实施组成分析。在使用3dap的情况下,能够排除在分析的区域中树脂成分或表面氧化等的影响,测定软磁性合金1、即内部区域2的组成。这是因为在利用3dap时,能够在软磁性合金1的内部设定小的区域(例如φ20nm
×
100nm的区域)并测定平均组成。
55.此外,在使用edx或eels(电子能量损失谱)对软磁性合金1的表面附近的截面进行线分析的情况下,内部区域2能够作为fe的浓度和co的浓度稳定的区域进行识别(参照图3a)。另外,例如,可以将内部区域2中的通过映射分析而得到的平均组成作为软磁性合金1的组成。在该情况下,映射分析使用edx或eels实施,此时的测定部位可以为软磁性合金1的在深度方向上距表面100nm以上的区域(相当于内部区域2),测定视场可以设为256nm
×
256nm左右的范围。
56.内部区域2的结晶结构(即软磁性合金1的结晶结构)可以为结晶质、纳米结晶、非晶质,更优选为非晶质。换言之,内部区域2的非晶质化度x(即软磁性合金1的非晶质化度x)优选为85%以上。非晶质化度x为85%以上的结晶结构为基本上由非晶质构成的结构、或由异质非晶构成的结构。在此,由异质非晶构成的结构是指在非晶质中存在微量结晶的结构。即,在本实施方式中,“非晶质的结晶结构”是指非晶质化度x为85%以上的结晶结构,也可以在满足该非晶质化度x的范围内包含结晶。
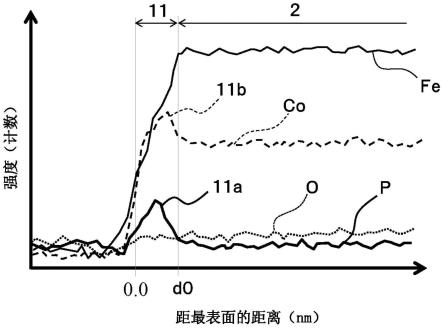
57.此外,在由异质非晶构成的结构的情况下,存在于非晶质中的结晶的平均结晶粒径优选为0.1nm以上10nm以下。另外,在本实施方式中,“纳米结晶”是指非晶质化度x低于85%、且平均结晶粒径为100nm以下(优选为3nm~50nm)的结晶结构,“结晶质”是指非晶质化度x低于85%、且平均结晶粒径超过100nm的结晶结构。
58.非晶质化度x可以通过使用xrd的x射线结晶结构解析来测定。具体而言,利用xrd对本实施方式的软磁性合金1进行2θ/θ测定,得到图2a所示的图。此时,衍射角2θ的测定范围设定为能够确认源自非晶质的晕圈的范围,例如优选设为2θ=30
°
~60
°
的范围。
59.接着,使用以下(2)式所示的洛伦兹函数,对图2a所示的图进行图形拟合。在该图形拟合中,优选将xrd实测的积分强度与使用洛伦兹函数算出的积分强度的误差设定在1%以内。通过该图形拟合,得到图2b所示的表示结晶性散射积分强度ic的结晶成分图案αc、表示非晶性散射积分强度ia的非晶成分图案αa、以及将它们合并的图案α
c+a
。然后,将在此得到的结晶性散射积分强度ic和非晶性散射积分强度ia导入以下(1)式,求出非晶质化度x。
60.x=100-(ic/(ic+ia)
×
100)
…
(1)
61.ic:结晶性散射积分强度
62.ia:非晶性散射积分强度
[0063][0064]
h:峰高度
[0065]
u:峰位置
[0066]
w:半值宽
[0067]
b:背景高度
[0068]
此外,非晶质化度x的测定方法并不限定于上述的使用xrd的方法,也可以通过ebsd(结晶方位解析)或电子射线衍射进行测定。
[0069]
(浓化部11)
[0070]
浓化部11含有fe、p、co等与内部区域2共同的构成元素,浓化部11中的与内部区域2共同的构成元素的合计含量以物质的量比换算计为50%以上,优选为80%以上。即,本实施方式中的浓化部11不是由磷酸盐处理等形成的涂层,而是由fe、p、co等构成的组合物的相,优选为从内部区域2连续的非晶质的金属相。
[0071]
如上所述,浓化部11由与内部区域2共同的元素构成,但在内部区域2和浓化部11中组成比不同。具体而言,在浓化部11中,p比内部区域2浓化。换言之,浓化部11存在p的浓度比上述的内部区域2高的p浓化区域11a,该p浓化区域11a覆盖内部区域2的外周缘的至少一部分。在软磁性合金1的截面中,浓化部11(即p浓化区域11a)相对于内部区域2的覆盖率没有特别限定,例如可以为50%以上,更优选为80%以上。
[0072]
另外,浓化部11中,优选co比内部区域2更浓化,优选浓化部11存在co浓化区域11b。该co浓化区域11b在从表面侧朝向内侧的软磁性合金1的深度方向(厚度方向)上,与p浓化区域11a重合存在。深度方向上的co浓化区域11b和p浓化区域11a的重合程度可以为一部分,也可以为全部,在部分重合的情况下,优选p浓化区域11a位于比co浓化区域11b靠浓化部11的表面侧。
[0073]
对于具有上述那样的特征的浓化部11,可以利用edx或eels(电子能量损失谱法)等进行分析,特别优选利用空间分辨率高的eels进行分析。
[0074]
例如,浓化部11的是否存在及其覆盖率可以通过如下方式确认,使用stem(扫描透射电子显微镜)或tem(透射电子显微镜)观察软磁性合金1的表面附近的截面,并在此时使用edx或eels实施映射分析。图5a所示的图像(eels图像)是eels的映射分析结果的一例。图5a的两个eels图像均为同一部位的测定结果,左侧的eels图像(p-l)表示p的分布,右侧的eels图像(co-l)表示co的分布。在该eels图像中,内部区域2能够作为co的浓度分布几乎不存在浓淡的区域进行识别。而且,可知在关于p的eels图像中,对比度在内部区域2的端缘变亮,p浓度比内部区域2高。该p浓度高的区域是p浓化区域11a。
[0075]
另外,可知在关于co的eels图像中,表示co的对比度在内部区域2的端缘变亮,co浓度比内部区域2高。可以确认该co浓度高的区域是co浓化区域11b,p浓化区域11a和co浓化区域11b在图5a中重合存在。
[0076]
由该映射分析确定的p浓化区域11a的平均厚度t1a优选为0.3nm以上。t1a的上限没有特别限定,例如可以为30nm以下。通过使t1a在该适当的范围内增厚,对于耐腐蚀性能够得到更好的结果。其中,平均厚度t1a优选改变测定视场在至少3个部位以上测定并计算p浓化区域11的厚度。
[0077]
co浓化区域11b的平均厚度t1b也可以为与上述t1a同等程度的范围,可以为t1a>t1b,也可以为t1a≤t1b。其中,浓化部11的厚度t1是p或/和co的检测强度高的区域的厚度,在不存在co浓化区域11b的情况下,t1=t1a。另外,在p浓化区域11a和co浓化区域11b完全重合的情况下,t1=t1a(t1a>t1b的情况)或t1=t1b(t1a≤t1b的情况)。
[0078]
如上所述,p浓化区域11a或co浓化区域11b有时厚度极薄,在确定p浓化区域11a和co浓化区域11b时,优选不仅利用映射分析,还并用线分析。图3a是示例沿着图1a所示的测定线lm进行线分析的结果的示意图,纵轴为各元素的检测强度(即特性x射线的强度),横轴为距最表面10的距离(深度)。如图3a所示,在线分析结果中,在fe或co的浓度稳定的内部区域2的端缘能够确认到p浓度变高的峰,存在该p的峰的部位为p浓化区域11a。换言之,在p浓化区域11a中,存在p浓度的最大值,根据上述峰的存在与否来确认是否存在p浓化区域11a。此外,在存在co浓化区域11b的情况下,如图3a所示,能够确认到以与p的峰重合的方式存在co的峰。
[0079]
另外,如上所述,浓化部11优选为金属相,浓化部11的相状态可以通过例如上述的线分析、映射分析、或使用stem或tem所附带的eels检测器的解析进行确认。例如,当对通过eels得到的谱图进行解析时,能够算出co浓化区域11b中的氧化物的co和金属co的比例,在金属co的比例大于氧化物的co的情况下,定义co浓化区域11为金属相。另外,在浓化部11的外侧存在氧化物层(后述的sb氧化层12、fe氧化层13、覆盖层20等)的情况下,在映射分析或线分析中,浓化部11中的氧的检测强度比氧化物层低。通过这种解析可知浓化部11为金属相。
[0080]
关于p浓化区域11a,利用强度比表示p的浓化程度,该强度比通过使用edx或eels的线分析算出。具体而言,将内部区域2中的p的检测强度设为c2
p
,将p浓化区域11a中的p的检测强度设为c11
p
,将c11
p
/c2
p
设为p浓化区域11a中的p强度比(浓化度)。该p强度比优选为1.3以上,更优选为1.5以上,进一步优选为2.0以上。p浓化度的上限值没有特别限定,例如可以设为20以下。
[0081]
另一方面,co浓化区域11b中的co浓化度定义为co浓化区域11b的co物质的量比(c11
co
)相对于内部区域2的co物质的量比(c2
co
)之比(c11
co
/c2
co
)。该co浓化度优选超过1.02,更优选超过1.20。此外,co浓化度的上限值没有特别限定,例如,可以为20以下。将未形成浓化部11的由内部区域2构成的软磁性合金作为基准合金时,存在co浓化度越高、本实施方式的软磁性合金1相对于该基准合金的耐腐蚀性越进一步提高的倾向。
[0082]
计算co浓化度所使用的c2
co
和c11
co
通过使用eels的成分分析进行测定。具体而言,c2
co
是内部区域2中检测到的co相对于fe和co的合计的物质的量比,通过eels谱图的解析算出。同样,c11
co
是在co浓化区域11b中检测到的co相对于fe和co的合计的物质的量比。即,各区域中的co的物质的量比以co/(fe+co)表示,为了排除杂质(制作测定试样时混入的元素等)的影响,将分母设为(fe+co)。
[0083]
此外,该分析中的分辨率优选设定为0.5nm以下,c2
co
优选在从软磁性合金1的最表面10向内部去深度为0.2μm以上的部位进行测定。另外,p强度比和co浓化度优选在至少5个部位以上的视场实施上述测定,作为其平均值算出。
[0084]
如上所述,软磁性合金1具有包括p浓化区域11a和/或co浓化区域11b的特征性的表层组织(浓化部11)。特别是本实施方式中,如图1a和图3a所示,p浓化区域11a位于最表面侧,构成软磁性合金1的最表面10。但是,也可以在p浓化区域11a的外侧存在其它的表层组织。
[0085]
例如,也可以如图1b所示的软磁性合金1a,以覆盖p浓化区域11a的表面侧的方式形成含有si或/和b的sb氧化层12。该sb氧化层12是选自si和b中的至少1种元素的浓度比内
部区域2高的区域,si或b中的任一方或si和b双方浓化。
[0086]
实际上,图5b为图1b所示的软磁性合金1a的eels图像的一例,图5b所示的4个eels图像均为同一部位的测定结果。在图5b的关于b的eels图像(上层右侧:b-k)中,能够确认在比p或co浓化的浓化部11靠表面侧,对比度变亮,该部位中的b的浓度比内部区域2和浓化部11高。在图5b的情况下,该b浓度高的区域为sb氧化层12。
[0087]
在内部区域2含有si或/和b的情况下,sb氧化层12有时在浓化部11的形成过程中生成,优选为非晶质的氧化物相。另外,sb氧化层12的平均厚度t2优选为0.5nm以上。t2的上限没有特别限定,例如能够设为30nm以下。
[0088]
另外,也可以在p浓化区域11a的外侧形成含有fe的fe氧化层13。该fe氧化层13有时在形成浓化部11的过程中与浓化部11一起生成,在fe氧化层13中,fe的浓度比浓化部11和内部区域2高。此外,如图1b所示,在sb氧化层12存在的情况下,fe氧化层13优选位于比sb氧化层12靠表面侧,进一步优选结晶化面积比比sb氧化层12高。
[0089]
实际上,在图5b的关于fe的eels图像(下层右侧:fe-l)中,可以确认在比sb氧化层12靠表面侧,对比度变亮,在软磁性合金1a的最表面存在fe浓度高的区域。在图5b中,该区域为fe氧化层13,fe氧化层13构成软磁性合金1a的最表面10。在本实施方式中,fe氧化层13的平均厚度t3优选为1nm以上。t3的上限没有特别限定,例如可以为50nm以下。
[0090]
图3b是模拟性地表示沿着图1b所示的测定线lma实施利用edx的线分析的结果的图。在存在sb氧化层12的情况下,如图3b所示,在比p的峰靠表面侧,观测到si或/和b的峰,并且以与该si或/和b的峰重合的方式,氧的检测强度升高。另外,在sb氧化层12的表面侧还存在fe氧化层13的情况下,在比si或/和b的峰靠表面侧能够确认到fe的峰。这样,sb氧化层12和fe氧化层13的存在与否可以通过使用edx或epma的线分析进行确认,除此之外,还能够通过图5b所示的映射分析等确认。
[0091]
另外,如图4a和图4b所示的软磁性合金1b,可以在p浓化区域11a的外侧形成绝缘性的覆盖层20。该覆盖层20是在形成浓化部11之后,通过涂布等表面处理形成的被膜,其平均厚度优选为5nm以上100nm以下,更优选为50nm以下。即,在形成覆盖层20的情况下,软磁性合金1b的最表面10由覆盖层20构成,位于比浓化部11、sb氧化层12、fe氧化层13靠软磁性合金1b的表面侧。实际上,图5c是图4a所示的软磁性合金1b的stem图像的一例。在该stem图像中,在软磁性合金1b的最表面10能够确认到对比度亮的区域,该区域是覆盖层20。
[0092]
这样,在软磁性合金1的表层组织中,除了浓化部11以外,还可以包括其它的层(sb氧化层12、fe氧化层13、覆盖层20等),但即使在存在其它的层的情况下,浓化部11也存在于与内部区域2相接的一侧。而且,从最表面到p浓化区域11a的垂线距离d1(参照图1b、图4b)优选为200nm以下,更优选为100nm以下,进一步优选为50nm以下。特别是在不存在覆盖层20,而由sb氧化层12或fe浓化区域13构成最表面10的情况下,上述垂线距离d1优选为30nm以下,更优选为20nm以下。
[0093]
此外,对浓化部11进行解析时的测定试样优选利用使用fib(聚焦离子束)的微采样法制作。例如,通过溅射在软磁性合金1的最表面10形成厚度30nm左右的用于加工时的表面保护的pt膜,然后,通过fib切出距最表面的深度约2μm左右的范围,得到薄片试样。然后,对该薄片试样进行加工,将与深度方向正交的方向的厚度减薄至20nm以下。可以将该经过薄膜化的试样用作tem或hrtem观察用的测定试样。
[0094]
以下,对本实施方式的软磁性合金1的制造方法进行说明。
[0095]
软磁性合金1的基体部(内部区域2)可以通过各种熔解法制造,特别优选通过将熔融金属(熔融液)骤冷的方法制造。这是因为通过骤冷容易得到非晶质的软磁性合金1的缘故。例如,薄带形状的软磁性合金1可以通过单辊法制造,粉末形状的软磁性合金1可以通过雾化法制造。以下,对通过单辊法得到软磁性合金薄带的方法、和通过作为雾化法一例的气体雾化法得到软磁性合金粉末的方法进行说明。
[0096]
利用单辊法时,首先,准备构成软磁性合金1的各元素的原料(纯金属等),以成为目的合金组成的方式称量。然后,将各元素的原料熔解,制作母合金。制作母合金时的熔解方法没有特别限定,例如,有通过高频加热使其在规定真空度的腔室内熔解的方法。
[0097]
接着,将上述的母合金加热使其熔解,得到熔融金属。熔融金属的温度可以考虑目的合金组成的熔点来设定,例如可以设为1200~1600℃。利用单辊法时,利用喷嘴等将该熔融金属供给至冷却后的旋转辊,朝向辊的旋转方向制造软磁性合金薄带。此时,通过控制辊的转速、喷嘴与辊的间隔、熔融金属的温度等,能够调整所得到的薄带的厚度。另外,辊的温度和转速可以设定为软磁性合金容易成为非晶质的条件,例如,辊温度优选为20~30℃,转速优选为20~30m/sec。此外,腔室内的气氛没有特别限定,例如,可以为大气气氛或不活泼气体气氛。
[0098]
利用气体雾化法时,与上述的单辊法同样,得到1200~1600℃的熔融金属后,在腔室内喷射该熔融金属,制作粉体。具体而言,从喷出口向腔室内的冷却部喷出熔融金属,此时,向喷出的滴加熔融金属喷射高压气体。通过高压气体的喷射,滴加熔融金属在腔室内飞散,然后,与冷却部(冷却水)碰撞,由此被骤冷固化,成为软磁性合金粉末。通过该气体雾化法得到的软磁性合金粉末的颗粒形状通常为球形,软磁性合金粉末的平均圆形度优选为0.8以上,更优选为0.9以上,进一步优选为0.95以上。
[0099]
作为高压气体,优选使用氮气、氩气、氦气等不活泼气体、或氨分解气体等还原性气体,喷射高压气体的压力优选为2.0mpa以上10mpa以下。另外,喷出的熔融金属的喷射量优选为0.5kg/min以上4.0kg/min以下。利用该气体雾化法时,可以根据高压气体的压力相对于熔融金属的喷射量的比率来调整软磁性合金粉末的粒径和形状。
[0100]
如上所述操作得到薄带状或粉末状的软磁性合金后,在规定压力状态下的氧浓度气氛中以低温对该软磁性合金进行热处理,由此,形成浓化部11。
[0101]
具体而言,热处理时的保持温度优选为软磁性合金不发生结晶化的温度,例如优选设为200℃~400℃。另外,温度保持时间优选设为0.5小时~3.0小时。加热炉内的氧浓度优选设为20ppm以上2000ppm以下,更优选设为100ppm以上1000ppm以下。另外,在加热炉内,优选如上所述管理氧浓度,并且导入氩气或氮气等不活泼气体并形成为正压,加热炉内的表压设为0.05kpa以上0.50kpa以下,在形成co浓化区域11b的情况下,更优选设为0.15kpa以上0.45kpa以下。其中,表压是指绝对压(将绝对真空设为0pa时的压力)减去了大气压后的压力。
[0102]
通过在这样的条件下进行热处理,在软磁性合金1的表层侧形成具有规定特征的p浓化区域11a(即浓化部11),在软磁性合金1含有co的情况下,有时也形成co浓化区域11a。另外,在软磁性合金1含有si或/和b的情况下,通过上述的热处理,有时形成sb氧化层12,根据热处理的条件,有时形成fe氧化层13。此外,在使软磁性合金1形成为结晶质或纳米结晶
的情况(即非晶质化度x低于85%的情况)下,也可以在实施用于形成上述浓化部11的热处理之前,实施用于控制结晶性的前工序热处理。
[0103]
在如图4a和图4b所示形成覆盖层20的情况下,可以在通过上述热处理形成浓化部11之后,实施磷酸盐处理、机械合金法、硅烷偶联处理、水热合成等被膜形成处理。作为所形成的覆盖层20的种类,可以举出磷酸盐、硅酸盐、钠钙玻璃、硼硅酸玻璃、铅玻璃、铝硅酸玻璃、硼酸盐玻璃、硫酸盐玻璃等。其中,作为磷酸盐,例如,可以举出磷酸镁、磷酸钙、磷酸锌、磷酸锰、磷酸化镉等,作为硅酸盐,可以举出硅酸钠等。在形成覆盖层20的情况下,能够期待包含软磁性合金1的磁芯中耐电压的提高等。
[0104]
通过以上的工序,得到具有规定的浓化部11的软磁性合金1。本实施方式的软磁性合金1能够适用于电感器等的线圈部件、滤波器、天线等的各种磁性部件,特别优选适用于电感器等的线圈部件的磁芯。此外,在包含软磁性合金1的磁芯中,可以含有树脂成分,也可以将软磁性合金1与其它的磁性颗粒混合而形成磁芯。
[0105]
(实施方式的总结)
[0106]
在本实施方式的软磁性合金1中,在具有含有fe和p的软磁性的合金组成的内部区域2的外侧形成有具有规定特征的p浓化区域11a。通过具有这种特征,能够抑制软磁性合金1浸水时生锈,提高耐腐蚀性。特别是通过p浓化区域11a中的p浓化度为1.5以上(更优选为2.0以上),能够进一步提高软磁性合金1的耐腐蚀性。
[0107]
另外,优选浓化部11以与p浓化区域11a重合的方式存在co浓化区域11b,通过形成co浓化区域11b,软磁性合金1的耐腐蚀性进一步提高。特别是通过使co浓化区域11b中的co浓化度超过1.20,能够进一步提高软磁性合金1的耐腐蚀性。
[0108]
另外,通过在非结晶化度为85%以上的非晶质的软磁性合金1中形成浓化部11,能够确保高的饱和磁通密度bs,并且进一步提高软磁性合金1的耐腐蚀性。
[0109]
以上,对本发明的实施方式进行了说明,但本发明并不限定于上述的实施方式,能够在本发明的范围内进行各种改变。
[0110]
实施例
[0111]
以下,基于具体的实施例更详细地说明本发明。但本发明并不限定于以下的实施例。其中,在下述所示的表中,标注
※
的试样编号为比较例。
[0112]
实验1
[0113]
在实验1中,通过气体雾化法制作软磁性合金粉末。在气体雾化中,设定为熔融金属的喷射温度:1500℃、熔融金属的喷射量1.2kg/min、高压气体的压力:7.0mpa、冷却水的水压:10mpa,得到体积基准的平均粒径(d50)处于15~30μm的范围内的软磁性合金粉末。然后,在表1所示的条件下对该软磁性合金粉末实施热处理,得到试样2~13的软磁性合金。另外,在实验1中,还制作未实施热处理的试样1的软磁性合金,以该试样1为基准,实施以下所示的评价。
[0114]
<软磁性合金粉末的组成和结晶结构>
[0115]
利用icp测定通过气体雾化法得到的软磁性合金粉末的组成。结果,确认在实验1的所有的试样中软磁性合金粉末(即内部区域2)具有满足组成式:(fe
0.7
co
0.3
)
0.82b0.11
p
0.02
si
0.03c0.01
cr
0.01
(原子数比:α=0.300、β=0、γ=0、a=0.110、b=0.020、c=0.030、d=0.010、e=0.010)的合金组成。另外,通过xrd对实验1的软磁性合金粉末实施x射
线结晶结构解析,结果确认在实验1的所有的试样中软磁性合金粉末(即内部区域2)是非晶质化度x为85%以上的非晶质。
[0116]
<表层组织的解析>
[0117]
对于实验1的各试样的软磁性合金,通过使用fib的微采样法取表层附近的薄片试样。然后,使用该薄片试样,利用tem-edx进行映射分析,调查有无浓化部11(p浓化区域11a和co浓化区域11b)。另外,利用tem-eels实施特定区域中的成分分析,测定浓化部11中的p浓化度和co浓化度。将表层组织的解析结果示于表1。此外,通过利用eels的解析,确认包含p浓化区域11a和/或co浓化区域11b的浓化部11为非晶质的金属相。
[0118]
<饱和磁通密度bs>
[0119]
使用振动试样型磁力计(vsm),在磁场1000ka/m的条件下测定各试样的软磁性合金的bs。将测定结果示于表1。对于该bs,将1.50t以上判断为良好,将1.70t以上判断为更好。
[0120]
<浸水试验>
[0121]
首先,在实施浸水试验前,使用各试样的软磁性合金制作磁芯样品。磁芯样品按照以下步骤制作。在软磁性合金100质量份中混合3质量份的环氧树脂,得到颗粒。然后,将该颗粒充填至模具中,以4ton/cm2的压力进行加压成型,得到外形内径高度1.0mm的环形的磁芯样品。
[0122]
为了评价上述所得到的磁芯样品的耐腐蚀性,实施浸水试验。在浸水试验中,将磁芯样品浸渍于自来水中,测量直到通过目测观察到生锈的时间(生锈时间)。在实验1中,将不实施热处理的试样1的生锈时间t1作为基准,评价各试样的耐腐蚀性。具体而言,在实验1中,将生锈时间相对于t1(试样1的生锈时间)低于1.0倍的样品判断为“f(不合格)”,将生锈时间相对于t1超过1.0倍且低于1.2倍的样品判断为“g(良好)”,将生锈时间相对于t1为1.2倍以上的样品判断为“vg(特别好)”。将上述的“f、g、vg”的3个等级的评价结果示于表1。
[0123]
【表1】
[0124][0125]
如表1所示可以确认,在以规定条件进行了热处理的试样3~13中形成有p浓化区域11a,生锈时间比试样1、2长。特别是确认在以比试样3高的表压进行了热处理的试样4~13中,以与p浓化区域11a重合的方式形成co浓化区域11b,结果耐腐蚀性比试样3进一步提高。根据这些结果可知,通过在软磁性合金的表面侧形成p浓化区域11a,能够确保高的bs,
并且能够提高相对于基准合金(试样1)的相对的耐腐蚀性。并且可知通过以与p浓化区域11a重合的方式形成co浓化区域11b,耐腐蚀性进一步提高。
[0126]
其中,在表1中省略了生锈时间的具体数值,但能够确认co浓化度越高,相对于基准的试样1的生锈时间越长,相对的耐腐蚀性越好的倾向。
[0127]
实验2
[0128]
在实验2中,改变合金组成,得到试样14~105的软磁性合金。将通过icp分析得到的各试样的合金组成示于表2~表7。
[0129]
具体而言,在表2所示的试样14~29中,制作满足组成式:(fe
1-α
co
α
)
0.82b0.11
p
0.02
si
0.03c0.01
cr
0.01
(原子数比:β=0、γ=0、a=0.110、b=0.020、c=0.030、d=0.010、e=0.010)、并且变更了co的原子数比α的软磁性合金。此外,试样22与表1的试样1相同,试样23与表1的试样11相同。
[0130]
另外,在表3所示的试样30~49中,制作将co、ni、x1的原子数比固定成α=0.300、β=0、γ=0、并且变更了非金属(b、p、si、c)和cr的原子数比的软磁性合金。
[0131]
另外,在表4所示的试样50~53中,制作满足组成式:(fe
(1-(0.3+β)
co
0.3
ni
β
)
0.82b0.11
p
0.02
si
0.03c0.01
cr
0.01
(原子数比:α=0.300、γ=0、a=0.110、b=0.020、c=0.030、d=0.010、e=0.010)、并且变更了ni的原子数比β的软磁性合金。
[0132]
另外,在表5~表7所示的试样54~105中,制作满足组成式((fe
0.7
co
0.3
)
0.975
x1
0.025
)
0.82b0.11
p
0.02
si
0.03c0.01
cr
0.01
(原子数比:α=0.300、β=0、γ=0.025、a=0.110、b=0.020、c=0.030、d=0.010、e=0.010)、并且变更了x1的元素种类的软磁性合金。
[0133]
此外,能够确认实验2的各软磁性合金的非晶质化度x均为85%以上。另外,在实验2中,按照每个合金组成,制作实施了规定的热处理的试样和未实施的试样,在表2~表7中,将实施了热处理的情况记作“y”,将未实施热处理的情况记作“n”。另外,实验2的热处理的条件设为保持温度:200℃、保持时间:1h、加热炉内的氧浓度:100ppm、加热炉内的表压:0.30kpa。
[0134]
另外,在实验2的各试样14~105中,也与实验1同样,实施了bs的测定和浸水试验。在实验2的浸水试验中,将以相同组成未实施热处理的试样的生锈时间tn作为基准,将实施了热处理的试样的生锈时间设为ty,将ty/tn<1.0的样品判断为“f(不合格)”,将1.0≤ty/tn<1.2的样品判断为“g(良好)”,将1.2≤ty/tn的样品判断为“vg(特别好)”。将评价结果示于表2~表7。
[0135]
[0136]
[0137]
[0138]
[0139]
[0140][0141]
如表2~表7所示,在实施了规定的热处理的试样中,得到了比未实施热处理的试样高的耐腐蚀性。根据该结果可知,在实验2所示的合金组成的范围内,通过形成具有规定特征的浓化部11(p浓化区域11a和co浓化区域11b),能够维持高的bs且提高耐腐蚀性。
[0142]
此外,作为对表2结果的补充,存在内部区域2中的co含量(即软磁性合金的co含量)越多,生锈时间越长的倾向。即,内部区域2中的co含量越多,作为绝对评价的耐腐蚀性越高。但是,如表2的试样29所示,内部区域2中的co含量高时,存在co浓化度反而容易降低的倾向。而且,就相对的耐腐蚀性的提高效果(即相对于基准合金的耐腐蚀性)而言,与试样
29相比,co浓化度高的其它的试样17、19、21、23、25、27得到良好的结果。即,根据该结果可以确认co浓化度越高,相对于基准合金(不实施用于形成浓化区域的热处理的试样)的耐腐蚀性的提高效果越高的倾向。
[0143]
实验3
[0144]
在实验3中,制造非晶质化度x为85%以上的非晶质的软磁性合金粉末(试样1、11)、非晶质化度x低于85%的纳米结晶的软磁性合金粉末(试样106、107)、非晶质化度x低于85%的结晶质的软磁性合金粉末(试样108、109),调查软磁性合金的结晶结构的不同对耐腐蚀性的影响。
[0145]
在实验3中,各试样的结晶结构通过前工序热处理进行控制。具体而言,实验3的试样1、11中,由于未实施前工序热处理,得到了非晶质的软磁性合金粉末。另外,实验3的试样106、107中,通过以保持温度:500℃实施前工序热处理,得到纳米结晶的软磁性合金粉末。另外,在实验3的试样108、109中,通过以保持温度:650℃实施前工序热处理,得到结晶质的软磁性合金粉末。其中,上述的前工序热处理中的其它条件设为升温速度:100℃/min、炉内气氛:ar气氛、加热炉内的表压:0.0kpa,在未形成浓化部11的状态下控制结晶结构。
[0146]
实验3的各试样中的软磁性合金的组成均为(fe
0.7
co
0.3
)
0.82b0.11
p
0.02
si
0.03c0.01
cr
0.01
,是相同的。另外,在实验3中,按照每个结晶结构,制作实施了用于形成浓化部11的热处理的试样和未实施的试样,在表8中,将实施了热处理的情况记作“y”,将未实施热处理的情况记作“n”。此外,在实施了前工序热处理的试样(107、109)中,在前工序热处理之后,实施用于形成浓化部11的热处理。另外,实验3中的该热处理的条件设为保持温度:200℃、保持时间:1.0h、加热炉内的氧浓度:100ppm、加热炉内的表压:0.3kpa。
[0147]
另外,在实验3中,也与实验2同样地实施了bs的测定和浸水试验。在实验3的浸水试验中,在相同的结晶结构中,将未实施热处理的试样的生锈时间tn作为基准,将实施了热处理的试样的生锈时间设为ty,将ty/tn<1.0的样品判断为“f(不合格)”,将1.0≤ty/tn<1.2的样品判断为“g(良好)”,将1.2≤ty/tn的样品判断为“vg(特别好)”。将实验3的评价结果示于表8。
[0148][0149]
如表8所示可知,即使是纳米结晶或结晶质的软磁性合金,也与非晶质的情况同样,在通过规定的热处理形成了p浓化区域11a和co浓化区域11b的试样107、109中,耐腐蚀性比未实施热处理的试样106、108提高。另外,对比表8所示的试样106~109的结果与试样1、11的结果,可知在软磁性合金为非晶质的情况下,相对于基准合金的生锈时间更长,相对的耐腐蚀性的提高效果特别好。
[0150]
实验4
[0151]
在实验4中,通过单辊法制作薄带形状的软磁性合金试样(试样110、111)。薄带制作的条件设为向辊喷射的熔融金属的温度:1300℃、辊温度:30℃、辊转速:25m/sec。腔室内设为大气气氛。通过上述条件得到的软磁性合金薄带的厚度为20~25μm,短边方向的宽度约为5mm,薄带的长度约为10m。
[0152]
另外,在实验4中,也与实验1同样地通过icp测定了试样110、111的合金组成,确认均满足组成式:(fe
0.7
co
0.3
)
0.82b0.11
p
0.02
si
0.03c0.01
cr
0.01
(原子数比:α=0.300、β=0、γ=0、a=0.110、b=0.020、c=0.030、d=0.010、e=0.010)。另外,通过xrd测定试样110、111的软磁性合金薄带的结晶结构,确认均是非晶质化度x为85%以上的非晶质。
[0153]
对于试样110的软磁性合金薄带,不实施热处理,实施表层组织的解析、bs的测定和浸水试验。另一方面,对于试样111的软磁性合金薄带,在表9所示的条件下实施热处理,然后,实施与试样110同样的评价。此外,在软磁性合金薄带的浸水试验中,通过将薄带切成任意的大小(长度约4cm
×
宽度约5mm),制备试验用样品,将薄带状的试验用样品浸渍于自来水中。实验4中的判定是否合格的方法与实验1相同。将实验4的各试样的评价结果示于表9。此外,在表9中还示出了与试样110、111相同合金组成的软磁性合金粉末的实验结果(实验1的试样1、11)。
[0154]
【表9】
[0155][0156]
如表9所示,能够确认即使在软磁性合金具有薄带形状的情况下,通过利用规定的热处理形成p浓化区域11a和co浓化区域11b,也能够维持高的bs且提高耐腐蚀性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1