一种锂离子电池负极用Fe掺杂的Si/C复合材料的制备方法与流程
一种锂离子电池负极用fe掺杂的si/c复合材料的制备方法
技术领域
1.本发明涉及一种锂离子电池负极用fe掺杂的si/c复合材料的制备方法,属于锂离子电池负极材料技术领域。
背景技术:
2.锂离子电池(libs)因具有高能量密度、长循环寿命和安全环保性而备受关注。为满足便携式电子设备和电动汽车不断增长的电力需求,libs的能量输出还需进一步提高。目前,libs应用最为广泛的负极材料为石墨,但其理论比容量仅有372mah/g,难以满足未来工业储能的需求。硅因具有高理论容量(4200mah/g)、低嵌/脱锂电位(0.2-0.3v vs li/ li
+ )和储量丰富等优点,被认为是很有发展潜力的负极候选材料之一。然而,硅负极在合金反应中的体积膨胀高达300%,易导致活性物质严重粉碎,充放电循环寿命短。形成的固体电解质界面膜(sei)造成阻抗增加、库仑效率降低和容量降低。此外,硅的本征电导率较低,阻碍了硅负极在充放电过程中的电荷转移。以上因素使硅负极材料的单独使用难以实现,限制了其在libs中的实际应用。
3.为了缓解锂离子在嵌入脱嵌过程中对硅负极的破坏,使用其他材料对晶体硅进行包覆是一种有效的方式。碳材料的结构比较稳定,可以作为si体积膨胀的缓冲基体,提高电子导电性并稳定硅基负极的sei层。硅材料作为活性成分,有助于高储锂容量。通过掺杂一定量的fe、ti或 ni等金属可以显著提升硅碳材料的首次效率。因此,制备具有高容量和高首效的硅碳复合材料成为libs负极材料的理想选择。
4.授权公告号为cn 104852027 b的发明专利,公开了一种具有三维笼状十二面体结构的si/c复合材料的制备方法,该专利以生长在对氨基苯甲酸功能化的纳米硅表面上的三维类沸石咪唑框架zif-8为前驱体,通过氮气保护下高温煅烧而后经盐酸处理制备具有三维笼状十二面体结构的si/c复合材料。该si/c复合材料的尺寸约为100~300nm,具有较大的比表面积和更多的孔洞结构,其作为锂离子电池的负极材料展示了较好的性能。但是,它在高温煅烧后还需要采用酸浸渍去除zno,步骤较为复杂;而且,目前的硅基材料仍存在脱嵌锂过程中体积膨胀严重、首次效率低、循环稳定性差等问题。
技术实现要素:
5.本发明要解决上述问题,从而提供一种锂离子电池负极用fe掺杂的si/c复合材料的制备方法。本发明的方法相对于现有技术,在煅烧后无须用酸处理,简化了操作,并且制得的负极材料,有效提高材料的容量发挥和循环寿命,并提高了首次效率,提升了电化学性能。
6.本发明解决上述问题的技术方案如下:一种锂离子电池负极用fe掺杂的si/c复合材料的制备方法,包括以下步骤:s1、将1.52~4.30质量份的硅纳米加入6.20~18.35质量份的n-乙烯基酰胺类聚合物溶液中,搅拌使之混匀,用甲醇/乙醇洗涤,干燥,得到粉末状中间产物si@n-乙烯基酰胺
类聚合物;s2、将3.34~4.25质量份si@n-乙烯基酰胺类聚合物、6.28~8.30质量份锌盐、1.85~2.60质量份铁盐、10.75~15.24质量份咪唑配体和100~200质量份溶剂加入反应容器中,搅拌使之混匀,静止放置20~23h;s3、继步骤s2,加入0.75~1.05质量份联吡啶,搅拌使之混匀,静止放置10~16h,分别用去离子水和甲醇/乙醇洗涤,干燥,得到十二面体结构的fe掺杂si-zif8前驱体;s4、将前驱体放置于管式炉中,密闭,通入惰性气体,排除内部空气,升温至900~1100℃,保温2~4h,得到fe掺杂的si/c复合材料。
7.金属有机骨架材料(mofs)是由金属离子与有机配体自组装杂化而成的一类具有多维网状结构的多孔材料。金属有机骨架材料在金属中心、配体结构、合成条件等方面具有极大的可调控性。向mofs结构中引入新的反应活性位点,如掺杂一定量的fe、ti或 ni等金属,可以显著提升硅碳材料的电化学性能。沸石咪唑酯骨架材料(zifs)是mofs中常见的一类,其中的金属离子主要是zn
2+
、co
2+
等,有机配体为咪唑或咪唑衍生物,通过咪唑环上的氮原子相连。通常将金属离子为zn
2+ 的沸石咪唑酯骨架材料称为zif-8。
8.本发明上述技术方案中,以zif-8为基底,因zn原子和fe原子的原子半径相近,采用基于“骨架金属竞争配位”的新策略,实现了zn和fe的快速掺杂,得到了一系列具有较好性能的硅碳负极材料。zif-8经过900℃以上的高温热解后,zn原子会以蒸汽形式蒸发形成纳米孔,丰富的孔隙结构能够提供足够的电极/电解质界面,促进电解质和si纳米颗粒之间的有效接触,加速电解液的渗透,减少锂离子的扩散距离,以改善锂离子的存储。
9.作为上述技术方案的优选,步骤s2中,所述的咪唑配体的制备方法为,将20~30质量份2-巯基-1-甲基咪唑、0.5~3质量份丙烯酸铁、100~120质量份乙醇、1~4质量份三乙胺搅拌均匀,除去乙醇。
10.作为上述技术方案的优选,步骤s2中,所述的锌盐选自乙酸锌、氯化锌、硝酸锌、硫酸锌中的至少一种。
11.作为上述技术方案的优选,步骤s2中,所述的铁盐选自硫酸铁、硝酸铁、氯化铁中的至少一种。
12.作为上述技术方案的优选,步骤s2中,所述的溶剂为甲醇和/或去离子水中。
13.作为上述技术方案的优选,步骤s3中,所述的联吡啶选自2,2
’‑
联吡啶、4,4
’‑
联吡啶中的至少一种。
14.作为上述技术方案的优选,步骤s1中,所述的n-乙烯基酰胺类聚合物为聚乙烯吡咯烷酮。
15.作为上述技术方案的优选,步骤s1中,所述搅拌使之混匀具体为,在室温下100~150r/min搅拌2~5h;步骤s2中,所述搅拌使之混匀具体为,在室温下150~200r/min搅拌1.5~4h;步骤s3中,所述搅拌使之混匀具体为,在室温下150~200r/min搅拌1~4h。
16.作为上述技术方案的优选,步骤s1中,所述干燥,具体为在60~90℃下真空干燥7~10h;步骤s3中,所述干燥,具体为在80~110℃下真空干燥5~8h。
17.作为上述技术方案的优选,所述的纳米硅的粒度分布范围为5~100nm。
18.综上所述,本发明具有以下有益效果:1、本发明在热解过程中,有机配体中的n原子进入碳环形成n掺杂碳,通过加入联
吡啶来补充氮源和碳源,有助于进一步提高碳骨架的电导率;2、采用本发明的方法,能够形成对si纳米粒子紧凑包覆的碳壳,可以适应硅粒子在循环过程中的大体积变化,且不使自身结构解体;包覆一层碳壳也有助于形成薄而致密的sei膜,抑制溶剂化锂离子的共嵌入,有效提高材料的容量发挥和循环寿命;3、本发明通过对咪唑配体进行改性,能够增强配体与fe之间的相容性;fe的引入能起到抑制惰性相生成的作用,提高首次效率;掺杂金属fe并未显著影响zif-8的主体结构,所得材料基本保留了zif-8多面体的形貌,并有助于提升硅碳材料的电化学性能;4、本发明前驱体的制备仅在室温下就可完成,该合成方法条件简单,绿色环保;5、本发明提供的fe掺杂硅碳复合材料实现了碳壳对si纳米粒子的包覆,能够基本保持原始的十二面体结构,缓解长期循环过程中体积变化导致的不良影响,用作锂离子电池的负极材料能展现出较高的容量;6、本发明制备的氮掺杂碳,有助于提高材料的电导率,在使用过程中能够减少对碳添加剂的需求,节约成本;7、本发明对si/c复合材料的合成方法具有借鉴意义,也为高性能的锂离子电池负极材料的发展提供了一种新的思路。
附图说明
19.图1为si/c材料的制备流程图;图2为实施例3制备的si/c材料扫描电镜图;图3为实施例3制备的si/c材料x射线衍射图;图4为实施例3制备的si/c材料充放电曲线图;图5为实施例3制备的si/c材料前100圈循环性能图。
具体实施方式
20.以下结合附图对本发明进行进一步的解释说明。
21.本具体实施方式仅仅是对本发明的解释,并不是对本发明的限制。本领域技术人员在阅读了本发明的说明书之后所做的任何改变,只要在权利要求书的范围内,都将受到专利法的保护。
22.实施例1s1、将1.52g硅纳米颗粒(10nm)加入6.20g聚乙烯吡咯烷酮溶液中,在室温下100r/min搅拌5h,用无水乙醇洗涤3次,再60℃下真空干燥10h,得到si@pvp中间产物粉末;s2、将3.34g中间产物、6.28g乙酸锌、1.85g硫酸铁、10.75g 改性咪唑配体和100ml甲醇加入反应容器中,在室温下200r/min搅拌1.5h,静止放置20h;s3、在反应容器中加入0.75份2,2
’‑
联吡啶,在室温下150r/min搅拌4h,静止放置10h,分别用去离子水和无水乙醇洗涤2次,在80℃下真空干燥8h,得到十二面体结构的fe掺杂的si-zif8前驱体;s4、将前驱体放置于管式炉中,密闭,通入氮气气体15min,排除内部空气,再升温至900℃,保温4h,然后自然冷却到室温,得到一种fe掺杂的si/c复合材料。
23.所述的改性咪唑配体的制备方法为,按将20质量份2-巯基-1-甲基咪唑、0.5质量
份丙烯酸铁、100质量份乙醇、1质量份三乙胺加入反应器中,在60℃下搅拌2h,然后减压蒸馏除去乙醇,得到所述的改性咪唑配体。
24.实施例2s1、将2.32g硅纳米颗粒(20nm)加入9.36g聚乙烯吡咯烷酮溶液中,在室温下120r/min搅拌3.5h,用无水乙醇洗涤4次,再70℃下真空干燥9h,得到si@pvp中间产物粉末;s2、将3.55g中间产物、6.42g氯化锌、2.05g硝酸铁、12.23g改性咪唑配体和125ml水加入反应容器中,在室温下180r/min搅拌2h,静止放置21h;s3、在反应容器中加入0.85份4,4
’‑
联吡啶,在室温下165r/min搅拌3h,静止放置12h,分别用去离子水和无水乙醇洗涤3次,在100℃下真空干燥6h,得到十二面体结构的fe掺杂的si-zif8前驱体;s4、将前驱体放置于管式炉中,密闭,通入氩气气体20min,排除内部空气,再升温至1000℃,保温3h,然后自然冷却到室温,得到一种fe掺杂的si/c复合材料。
25.所述的改性咪唑配体的制备方法为,按将23质量份2-巯基-1-甲基咪唑、1.3质量份丙烯酸铁、105质量份乙醇、2质量份三乙胺加入反应器中,在65℃下搅拌2.5h,然后减压蒸馏除去乙醇,得到所述的改性咪唑配体。
26.实施例3s1、将3.57g硅纳米颗粒(50nm)加入14.27g聚乙烯吡咯烷酮溶液中,在室温下130r/min搅拌3h,用无水乙醇洗涤6次,再80℃下真空干燥8h,得到si@pvp中间产物粉末;s2、将3.72g中间产物、7.65g硝酸锌、2.38g氯化铁、13.06g改性咪唑配体和150ml甲醇加入反应容器中,在室温下160/min搅拌3h,静止放置22h;s3、在反应容器中加入0.95份2,2
’‑
联吡啶,在室温下185r/min搅拌2h,静止放置14h,分别用去离子水和无水乙醇洗涤4次,在110℃下真空干燥5h,得到十二面体结构的fe掺杂的si-zif8前驱体;s4、将前驱体放置于管式炉中,密闭,通入氮气气体20min,排除内部空气,再升温至950℃,保温2.5h,然后自然冷却到室温,得到一种fe掺杂的si/c复合材料。
27.所述的改性咪唑配体的制备方法为,按将27质量份2-巯基-1-甲基咪唑、2.2质量份丙烯酸铁、110质量质量份乙醇、3质量份三乙胺加入反应器中,在70℃下搅拌3h,然后减压蒸馏除去乙醇,得到所述的改性咪唑配体。
28.实施例4s1、将4.30g硅纳米颗粒(100nm)加入18.35g聚乙烯吡咯烷酮溶液中,在室温下150r/min搅拌2h,用无水乙醇洗涤5次,再90℃下真空干燥7h,得到si@pvp中间产物粉末;s2、将4.25g中间产物、8.30g硫酸锌、2.60g氯化铁、15.24g改性咪唑配体和200ml甲醇加入反应容器中,在室温下150/min搅拌4h,静止放置23h;s3、在反应容器中加入1.05份4,4
’‑
联吡啶,在室温下200r/min搅拌1h,静止放置16h,分别用去离子水和无水乙醇洗涤5次,在90℃下真空干燥7h,得到十二面体结构的fe掺杂的si-zif8前驱体;s4、将前驱体放置于管式炉中,密闭,通入氩气气体25min,排除内部空气,再升温至1100℃,保温2h,然后自然冷却到室温,得到一种fe掺杂的si/c复合材料。
29.所述的改性咪唑配体的制备方法为,将30质量份2-巯基-1-甲基咪唑、3质量份丙
烯酸铁、120质量份乙醇、4质量份三乙胺加入反应器中,在75℃下搅拌4h,然后减压蒸馏除去乙醇,得到改性咪唑配体。
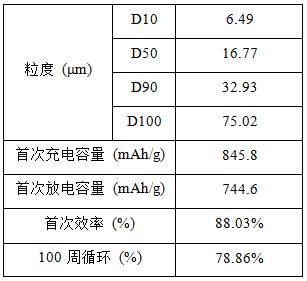
30.性能测试方法如下:粒度测试方法为,取0.2g样品超声分散在20ml纯水中,超声处理的样品加入进样系统,遮光度为12-15%,待稳定后通过软件直接读取d10、d50、d90、d100值。
31.电性能测试方法为,组装扣电后使用蓝电测试柜进行。
32.图2为实施例3制备的si/c材料扫描电镜图;从图2可以看出,si/c复合材料基本保持了zif-8原始的十二面体结构,高温下咪唑配体分解导致其表面变得粗糙,尺寸约为250nm;图3为实施例3制备的si/c材料x射线衍射图;从图3可以看出,si/c复合材料已成功合成;图4为实施例3制备的si/c材料充放电曲线图;从图4可以看出,si/c复合材料具有良好的充放电电压平台和高容量,首次充电容量达到845.8 mah/g,首次放电容量达到744.6 mah/g;图5为实施例3制备的si/c材料前100圈循环性能图;从图5可以看出,si/c复合材料在循环初期容量衰减较快,但在之后的循环中容量保持稳定,经过100周循环后容量保持率仍达到78.86%。
33.表1为实施例3制备的si/c材料测试数据。
34.表1
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1