一种碳气凝胶及其制备方法与流程
1.本发明涉及碳气凝胶技术领域,具体涉及一种超级电容器电极用碳气凝胶及其制备方法。
背景技术:
2.碳气凝胶是一种由纳米碳颗粒相互连结而成的纳米级多孔材料,其基本特点为孔隙率高以及比表面积高,因其具有高导电率、可直接成型等特点,是制备超级电容器的理想电极材料之一,也是当前研究的热点。在传统碳气凝胶的制备过程中,通常采用溶胶凝胶法制成有机湿凝胶,再经超临界co2干燥转化为有机气凝胶,最后在惰性气体保护下将有机气凝胶高温碳化而成。其中,超临界干燥是溶胶凝胶法制备碳气凝胶的重要步骤,这是由于超临界干燥可以消除气体界面,有效保持凝胶孔结构,从而保证湿凝胶碳化后保持三维网络结构。但是超临界干燥成本高、过程复杂、产率低,且具有一定的危险性,严重阻碍了碳气凝胶的产业化进程。
3.针对超临界干燥有机湿凝胶工艺存在的具体问题,开发可替代超临界干燥的其他干燥工艺来干燥有机湿凝胶成为当前的研究热点之一。冷冻干燥被认为是替换超临界干燥工艺的优选方法。中国专利cn113387344a中,通过添加环氧氯丙烷辅助形成前驱体凝胶并采用冷冻干燥制备出碳气凝胶,工艺流程如下:首先将碳纳米管分散于去离子水中,制得碳纳米管分散液;然后向所述碳纳米管分散液中加入间苯二酚、甲醛和碳酸钠,同时滴入环氧氯丙烷,制得水凝胶前驱体溶胶后密封放置形成前驱体凝胶;再将所述前驱体凝胶进行溶剂置换,并冷冻干燥,制得酚醛包覆的碳纳米管气凝胶;最后对所述酚醛包覆的碳纳米管气凝胶高温煅烧,从而制得碳气凝胶。中国专利cn113120879a提供了一种按冷冻干燥工艺制备出兼具优良的电磁屏蔽性能和生物传感性能的碳气凝胶材料的方法,通过将纳米纤维素-氧化石墨烯水分散液与聚甲基丙烯酸甲酯溶液混合,得到纳米纤维素稳定的水包油型乳液;然后冷冻干燥,得到纳米纤维素-氧化石墨烯/聚甲基丙烯酸甲酯复合气凝胶;最后退火处理得到碳气凝胶材料。由于冷冻干燥需要在-50℃的低温以及30pa以下的极高真空度下才能进行,干燥效率及产率均很低,这是因为溶剂挥发的速率很慢,干燥时间一般不低于48h,干燥的物料填充比也很少,使得干燥成本高,难以大规模生产。
4.常压干燥是另一种替换超临界干燥过程制备碳气凝胶的优选方法。中国专利cn111498828a提供了一种利用常压干燥制备碳气凝胶的方法,通过在反应单体和催化剂体系中引入f127高分子聚合物增强剂。利用溶胶-凝胶法制成湿凝胶并进行老化处理,用低表面张力溶剂置换出湿凝胶中的水分,再置于空气中常压干燥即得有机气凝胶,最后将有机气凝胶碳化即得碳气凝胶。与加入p123相比,加入f127高分子聚合物增强剂制备出的碳气凝胶的孔径更大,材料的比表面积更高(可达801.1m2/g),从而更有利于电解液的传输以及材料电容性能的进一步提升。不过,由于制备出的材料孔径达到2μm以上,将这样的材料用于超级电容器时,较大的孔隙不仅会吸纳过多的电解液,增加超级电容器制造成本,而且会显著降低超级电容器电极的压实密度,最终导致超级电容器产品的体积能量密度偏低。中
国专利cn113860284a公开了一种在常压下快速加热干燥制备碳气凝胶的方法,先将类石墨微晶碳纳米材料、酚、甲醛、催化剂和去离子水混合成均匀悬浮液,密封静置实现凝胶化;然后将湿凝胶转移到烘箱进行老化处理,在管式炉中进行干燥,最后碳化得到碳气凝胶。这种方法通过直接加热干燥有机凝胶,过程简单,大大缩短了制备时间,也有利于将干燥与碳化过程集成为一个加热的连续化操作过程。但是,由于该方法在制备碳气凝胶过程中加入了类石墨微晶碳纳米材料,导致制备出的碳气凝胶中包含较多的类石墨微晶碳纳米材料杂质,这些杂质会堵塞碳气凝胶的一些微孔结构,导致碳气凝胶的离子传输能力和循环稳定性不佳,因此难以应用于超级电容器电极中。
5.中国专利cn112645302a公开了一种碳气凝胶的产业化生产制备方法。工艺流程为:将间苯二酚和间苯三酚的混合物与甲醛和去离子水混合,再加入脂环胺类混合均匀并在56℃进行凝胶反应,得到中间产物;对中间产物依次进行老化、干燥和分步碳化处理,得到碳气凝胶。这种方法解决了酸洗、溶剂交换过程中产生的废液量大和超临界干燥成本高的问题,实现了碳气凝胶的产业化生产。不过,由于间苯二酚和间苯三酚的混合物与甲醛发生凝胶化反应的时间很长,导致碳气凝胶的生产效率较低。
6.综上所述,碳气凝胶的制备工艺仍有较大提升空间。
技术实现要素:
7.本发明的目的在于:
8.(1)针对现有溶胶凝胶法制备碳气凝胶工艺流程复杂、有机湿凝胶干燥过程具有危险性、生产成本高、产率低以及生产出的碳气凝胶材料密度低的固有问题,提供一种高比表面积、高孔隙率且具有三维连通孔洞结构的碳气凝胶材料及其制备方法;
9.(2)提供一种压实密度高的超级电容器用电极材料;
10.(3)提供一种兼具高体积能量密度和高功率密度的超级电容器。
11.为实现上述目的,本发明采用的技术方案如下:
12.一方面,本发明提供一种碳气凝胶的制备方法,包括以下步骤:
13.s1、分别配制铝盐微乳液和氨水微乳液;
14.s2、将s1中的铝盐微乳液和氨水微乳液混合,然后将混合溶液进行静电纺丝,得到纳米氧化铝前驱体;
15.s3、将s2中的纳米氧化铝前驱体和可溶性有机碳源加入溶剂中,搅拌成浆料,然后干燥得到有机碳源/纳米氧化铝前驱体复合物;
16.s4、将s3中的有机碳源/纳米氧化铝前驱体复合物高温碳化,得到所述碳气凝胶。
17.进一步地,步骤s1中,所述铝盐微乳液和氨水微乳液的制备方法包括:将去离子水、表面活性剂和油混合得到微乳液,并将所述微乳液分成两份,其中一份加入铝盐,另一份加入氨水,充分搅拌后分别得到铝盐微乳液和氨水微乳液。其中,“将所述微乳液分成两份”可以随意分配(平分或者不平分均可),根据二者随意分配的体积,分别添加合适用量的铝盐和氨水,从而得到特定浓度的铝盐微乳液和氨水微乳液。
18.优选地,所述去离子水、表面活性剂和油的质量比为(45-65):(10-20):(30-45)。通过控制去离子水、表面活性剂和油的质量比,形成油包水型微乳液,确保铝盐微乳液和氨水微乳液经机械混合后不会发生破乳反应。
19.在本发明的具体实施例中,所述去离子水、表面活性剂和油的质量比为50:10:40、50:20:30、60:10:30。
20.优选地,所述铝盐微乳液的浓度为0.015-0.05mol/l,所述氨水微乳液的浓度为0.045-0.15mol/l。
21.更优选地,所述铝盐微乳液的浓度为0.015-0.03mol/l,所述氨水微乳液的浓度为0.08-0.1mol/l。
22.在本发明中,所述“微乳液的浓度”是指溶质分布于液相总体积中的摩尔浓度,例如,所述“铝盐微乳液的浓度”表示铝盐分布于微乳液总体积中的摩尔浓度。
23.在本发明的具体实施例中,所述铝盐微乳液的浓度为0.015mol/l、0.016mol/l、0.02mol/l、0.027mol/l、0.029mol/l、0.03mol/l、0.04mol/l、0.05mol/l。
24.在本发明的具体实施例中,所述氨水微乳液的浓度为0.045mol/l、0.05mol/l、0.06mol/l、0.07mol/l、0.08mol/l、0.081mol/l、0.087mol/l、0.09mol/l、0.096mol/l、0.1mol/l、0.15mol/l。
25.优选地,所述表面活性剂选自十二烷基硫酸钠(sds)、十二烷基三甲基溴化铵(dtab)、十六烷基三甲基溴化铵(ctab)、双(2-乙基己基)琥珀酸酯磺酸钠(aot)中的一种或多种;所述油选自环己烷、正庚烷、正辛烷、正壬烷和正癸烷中的一种或多种。
26.进一步地,所述铝盐选自alcl3、al2(so4)3、al(no3)3中的一种或多种。
27.进一步地,所述氨水微乳液的ph为10-11,例如:10、10.2、10.6、10.7、10.9、11。
28.在本发明中,步骤s2中,将混合溶液进行静电纺丝后,需要对静电纺丝所得产物进行定长切割(即周期性剪断)处理,确保纳米氧化铝前驱体堆积均匀,因此只要能实现定长切割的静电纺丝方式均能用于本发明。
29.优选地,步骤s2中,所述静电纺丝为脉冲式静电纺丝。更优选地,步骤s2中,所述静电纺丝为脉冲频率为100-200hz的脉冲式静电纺丝,例如,所述脉冲频率为100hz、120hz、140hz、150hz、160hz、170hz、180hz、190hz、200hz。
30.优选地,所述纳米氧化铝前驱体为长度100-500μm的絮状产物。如果纳米氧化铝前驱体长度过长,易于发生卷曲缠绕,难以将纳米氧化铝前驱体和可溶性有机碳源在溶剂中混合均匀;如果纳米氧化铝前驱体长度过短,自然堆积后形成的孔隙率会偏小,导致制备出的碳气凝胶孔隙率偏低。
31.更优选地,所述纳米氧化铝前驱体为长度160-330μm的絮状产物。
32.在本发明的具体实施例中,所述纳米氧化铝前驱体的长度为100μm、150μm、160μm、200μm、202μm、220μm、230μm、300μm、330μm、400μm、500μm。
33.进一步地,步骤s2中,铝盐微乳液和氨水微乳液按照(4-7):(3-6)的质量比例进行混合。
34.在本发明的具体实施例中,铝盐微乳液和氨水微乳液的质量比为1:1、11:9、6:4、6:5。
35.将铝盐微乳液和氨水微乳液混合是指常规的机械混合,例如搅拌,由于微乳液为油包水型,在机械搅拌混合过程中不会发生破乳反应;然后将混合溶液进行静电纺丝,静电纺丝过程会使混合溶液中的溶剂(包括去离子水和油)发生挥发,从而改变了铝盐微乳液和氨水微乳液的热力学稳定性,在这个过程中,微乳液液滴间的相平衡受到破坏,产生剧烈碰
撞,最终引起破乳反应。即在静电纺丝过程中,双相微乳液发生液滴碰撞破乳进而产生化学反应al
3+
+nh3·
h2o
→
al(oh)3,得到纳米氧化铝前驱体,即氢氧化铝。
36.进一步地,在静电纺丝过程中得到长度为100-500μm的絮状产物后,用去离子水清洗絮状产物3次,除去絮状产物表面的杂质,然后80℃烘干得到所述纳米氧化铝前驱体。
37.进一步地,步骤s3中,所述纳米氧化铝前驱体与可溶性有机碳源的质量比为(2-5):(4-8)。由此,使得可溶性有机碳源饱和填充在纳米氧化铝前驱体堆积的孔隙中。
38.在本发明中,所述可溶性有机碳源为含碳量不低于60%的可溶性有机碳源。优选地,所述可溶性有机碳源为酚醛树脂、环氧树脂、聚酯树脂、聚酰胺树脂、聚乙烯、聚苯乙烯、石油沥青、焦油沥青中的一种或多种。
39.优选地,步骤s3中,所述溶剂为碳数1-4的短链醇类溶剂,更优选为正丁醇、甲醇、无水乙醇、1-丙醇、2-丙醇、乙二醇、二乙二醇、1,2-丙二醇、1,3-丙二醇、1,4-丁二醇、丙三醇中的一种或多种。
40.优选地,步骤s3中,所述溶剂与固体物(包括纳米氧化铝前驱体和可溶性有机碳源)的质量比为(2-6):(5-8)。
41.在本发明的具体实施例中,所述溶剂与固体物的质量比为3:5、4:7、5:8、4:6.5。
42.优选地,步骤s3中,所述浆料的粘度为8000-16000cp,例如:8000cp、10000cp、11300cp、12500cp、12900cp、13800cp、14000cp、15000cp、16000cp。在本发明中,浆料的粘度由溶剂的含量决定,溶剂含量越高,浆料粘度越低。
43.更优选地,步骤s3中,所述浆料的粘度为10000-14000cp。
44.优选地,步骤s3中,所述干燥的温度为60-150℃,干燥时间为2-10h,优选干燥时间为2-4h。
45.在本发明中,纳米氧化铝前驱体经过干燥,随机堆叠成具有孔隙的产物,酚醛树脂填充在所述孔隙中,形成可溶性有机碳源/纳米氧化铝前驱体复合物。
46.进一步地,步骤s4中,所述高温碳化温度为700-900℃,升温速率为2-4℃/min,保温时间为4-6h。
47.进一步地,所述高温碳化在回转炉中进行;高温碳化过程中通入高纯保护性气体,所述保护性气体为氮气、氩气、氦气和氖气中的一种或多种。
48.进一步地,在所述高温碳化之后,还包括:将高温碳化得到的产物用酸清洗,干燥后得到所述碳气凝胶。优选地,所述酸为1mol/l浓度的盐酸,酸清洗次数为3次,所述干燥为150℃真空干燥。进行酸清洗的目的是除去纳米氧化铝前驱体,即酸与氢氧化铝发生反应,从而得到碳气凝胶产物。
49.第二方面,本发明提供一种由上述制备方法得到的碳气凝胶,所述碳气凝胶的比表面积不低于840g/m2,孔容积不低于0.375cm3/g。
50.第三方面,本发明提供一种包含上述碳气凝胶的超级电容器用电极,所述电极的压实密度不低于0.886g/cm3。
51.第四方面,本发明提供一种包含上述电极的超级电容器。由本发明提供的碳气凝胶材料制备成电极,并组装成φ22
×
45焊针式超级电容器,所得超级电容器兼具高体积能量密度及高功率密度特性。
52.本发明的有益效果在于:
53.(1)将双组分微乳液反应物在静电纺丝条件下破乳反应,获得长度为100-500μm的絮状纳米氧化铝前驱体,为制备具有三维连通孔洞结构的碳气凝胶提供了合适的模板;
54.(2)将纳米氧化铝前驱体与可溶性有机碳源液相混合并干燥,获得可溶性有机碳源填充纳米氧化铝前驱体堆积孔隙的中间产物(有机碳源/纳米氧化铝前驱体复合物),经碳化后除去模板纳米氧化铝前驱体得到碳气凝胶,与现有技术相比,本发明碳气凝胶制备成超级电容器用电极后,电极材料压实密度高,有利于提高超级电容器的体积能量密度;
55.(3)与溶胶凝胶法制备碳气凝胶的工艺流程相比,本发明有效避免了有机湿凝胶的形成及干燥过程,制备方法简单、生产效率高,非常有利于生产企业推广使用。
56.术语定义
57.除非明确地说明与此相反,否则,本发明引用的所有范围包括端值。
58.本发明使用的术语“至少一种”来描述本发明所描述的要素和组分。这样做仅仅是为了方便,并且对本发明的范围提供一般性的意义。这种描述应被理解为包括一种或至少一种,并且该单数也包括复数,除非明显地另指他意。
59.本发明中的数字均为近似值,无论有否使用“大约”或“约”等字眼。数字的数值有可能会出现1%、2%、5%、7%、8%、10%等差异。每当公开一个具有n值的数字时,任何具有n+/-1%,n+/-2%,n+/-3%,n+/-5%,n+/-7%,n+/-8%或n+/-10%值的数字会被明确地公开,其中“+/
‑”
是指加或减,并且n-10%到n+10%之间的范围也被公开。
60.除非另外说明,应当应用本发明所使用的下列定义。出于本发明的目的,化学元素与元素周期表cas版,和1994年第75版《化学和物理手册》一致。此外,有机化学一般原理可参考"organic chemistry",thomas sorrell,university science books,sausalito:1999,和"march's advanced organic chemistry"by michael b.smith and jerry march,john wiley&sons,new york:2007中的描述,其全部内容通过引用并入本发明。
61.除非另行定义,否则本发明所用的所有科技术语的含义与本发明所属领域的普通技术人员通常理解的一样。尽管与本发明所描述的方法和材料类似或等同的方法和材料也可用于本发明实施方案的实施或测试中,但是下文描述了合适的方法和材料。本发明提及的所有出版物、专利申请、专利以及其他参考文献均以全文引用方式并入本发明,除非引用具体段落。如发生矛盾,以本说明书及其所包括的定义为准。此外,材料、方法和实施例仅是例示性的,并不旨在进行限制。
附图说明
62.图1为实施例1中碳气凝胶的扫描电镜图;
63.图2为本发明对比例2中碳气凝胶的扫描电镜图。
具体实施方式
64.以下所述仅为本发明的较佳实施方式而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
65.实施例1
66.s1、按照50:10:40的质量比配制去离子水/sds/环己烷的微乳液,将该微乳液分成两份,其中一份加入alcl3,充分搅拌得到0.027mol/l的alcl3微乳液;另一份微乳液中加入
氨水,充分搅拌后得到0.081mol/l、ph=10.2的氨水微乳液;
67.s2、将s1中的alcl3微乳液和氨水微乳液按照1:1的质量比加入反应釜中搅拌均匀,将混合均匀的微乳液在150hz脉冲频率下进行脉冲式静电纺丝,获得长度为230μm的絮状产物,用去离子水将絮状产物清洗3次,除去絮状产物表面的杂质,然后80℃烘干得到纳米氧化铝前驱体;
68.s3、将s2中的纳米氧化铝前驱体和酚醛树脂按1:2的质量比加入正丁醇中,其中正丁醇与固体物(包括纳米氧化铝前驱体和酚醛树脂)的质量比为4:6.5,然后搅拌成粘度为12500cp的浆料,经120℃干燥3h后,得到酚醛树脂/纳米氧化铝前驱体复合物;
69.s4、将s3中的酚醛树脂/纳米氧化铝前驱体复合物加入回转炉中,通入高纯氩气为保护气,以3℃/min的加热速率升温至800℃并保温5h,得到含有纳米氧化铝前驱体的碳气凝胶粗产物;将该粗产物用1mol/l盐酸清洗3次,除去纳米氧化铝前驱体,最后150℃真空干燥得到所述碳气凝胶。
70.实施例2
71.s1、按照50:10:40的质量比配制去离子水/dtab/正庚烷的微乳液,将该微乳液分成两份,其中一份加入al2(so4)3,充分搅拌得到0.015mol/l的al2(so4)3微乳液;另一份微乳液中加入氨水,充分搅拌后得到0.09mol/l、ph=10.7的氨水微乳液;
72.s2、将s1中的al2(so4)3微乳液和氨水微乳液按照11:9的质量比加入反应釜中搅拌均匀,对混合均匀的微乳液在120hz脉冲频率下进行脉冲式静电纺丝,获得长度为330μm的絮状产物,用去离子水将絮状产物清洗3次,除去絮状产物表面的杂质,然后80℃烘干得到纳米氧化铝前驱体;
73.s3、将s2中的纳米氧化铝前驱体和酚醛树脂按1:2的质量比加入正丁醇中,其中正丁醇与固体物(包括纳米氧化铝前驱体和酚醛树脂)的质量比为5:8,然后搅拌成粘度为11300cp的浆料,经120℃干燥3h后,得到酚醛树脂/纳米氧化铝前驱体复合物;
74.s4、同实施例1的s4,得到所述碳气凝胶。
75.实施例3
76.s1、按照50:10:40的质量比配制去离子水/ctab/正辛烷的微乳液,将该微乳液分成两份,其中一份加入al2(so4)3,充分搅拌得到0.016mol/l的al2(so4)3微乳液;另一份微乳液中加入氨水,充分搅拌后得到0.096mol/l、ph=10.9的氨水微乳液;
77.s2、将s1中的al2(so4)3微乳液和氨水微乳液按照6:4的质量比加入反应釜中搅拌均匀,对混合均匀的微乳液在180hz脉冲频率下进行脉冲式静电纺丝,获得长度为160μm的絮状产物,用去离子水将絮状产物清洗3次,除去絮状产物表面的杂质,然后80℃烘干得到纳米氧化铝前驱体;
78.s3、将s2中的纳米氧化铝前驱体和酚醛树脂按1:2的质量比加入正丁醇中,其中正丁醇与固体物(包括纳米氧化铝前驱体和酚醛树脂)的质量比为4:7,然后搅拌成粘度为13800cp的浆料,经120℃干燥3h后,得到酚醛树脂/纳米氧化铝前驱体复合物;
79.s4、同实施例1的s4,得到所述碳气凝胶。
80.实施例4
81.s1、按照50:10:40的质量比配制去离子水/aot/正癸烷的微乳液,将该微乳液分成两份,其中一份加入al(no3)3,充分搅拌得到0.029mol/l的al(no3)3微乳液;另一份微乳液
中加入氨水,充分搅拌后得到0.087mol/l、ph=10.6的氨水微乳液;
82.s2、将s1中的al(no3)3微乳液和氨水微乳液按照6:5的质量比加入反应釜中搅拌均匀,对混合均匀的微乳液在160hz脉冲频率下进行脉冲式静电纺丝,获得长度为202μm的絮状产物,用去离子水将絮状产物清洗3次,除去絮状产物表面的杂质,然后80℃烘干得到纳米氧化铝前驱体;
83.s3、将s2中的纳米氧化铝前驱体和酚醛树脂按1:2的质量比加入正丁醇中,其中正丁醇与固体物(包括纳米氧化铝前驱体和酚醛树脂)的质量比为3:5,然后搅拌成粘度为12900cp的浆料,经120℃干燥3h后,得到酚醛树脂/纳米氧化铝前驱体复合物;
84.s4、同实施例1的s4,得到所述碳气凝胶。
85.对比例1
86.s1、将间苯二酚和甲醛按1:2的摩尔比加入无水乙醇中搅拌均匀,加入催化剂碳酸钠形成混合溶液,将该混合溶液置于60℃恒温水浴中放置7天,形成有机湿凝胶;
87.s2、向s1中的有机湿凝胶中加入体积分数为5%的乙酸溶液,浸渍老化3天,得到老化湿凝胶;
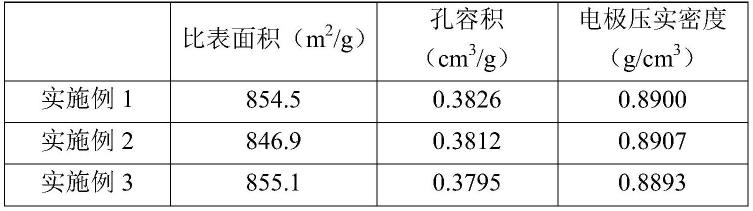
88.s3、用无水乙醇重复置换s2所得老化湿凝胶6次,使老化湿凝胶中的溶剂几乎全部替换为无水乙醇,得到有机气凝胶前驱体;
89.s4、用将s3中的有机气凝胶前驱体转移到干燥釜中,通入超临界co2干燥48h,得到有机气凝胶;
90.s5、将s4中的有机气凝胶加入回转炉中,通入高纯氩气为保护气,以3℃/min的加热速率升温至800℃并保持5h,得到碳气凝胶。
91.对比例2
92.s1、将间苯二酚和甲醛按1:2的摩尔比加入无水乙醇中搅拌均匀,加入碳酸钠和表面活性剂p123(聚醚:聚氧乙烯-聚氧丙烯-聚氧乙烯)继续搅拌形成混合溶液。将混合溶液置于60℃恒温水浴中放置7天,形成有机湿凝胶;
93.s2、向s1中的有机湿凝胶中加入体积分数为5%的三氟乙酸溶液,浸渍老化3天,得到老化湿凝胶;
94.s3、用丙酮重复置换s2中的老化湿凝胶6次,使老化湿凝胶中的溶剂几乎全部替换为丙酮,得到有机气凝胶前驱体;
95.s4、将s3中的有机气凝胶前驱体置于室温常压下放置7天,使有机气凝胶前驱体内的溶剂丙酮自然挥发完全,得到有机气凝胶;
96.s5、将s4中的有机气凝胶加入回转炉中,通入高纯氩气为保护气,以3℃/min的加热速率升温至800℃并保持5h,得到碳气凝胶。
97.性能评价
98.1、碳气凝胶性能测试
99.选取实施例1及对比例2的碳气凝胶样品作为本发明及现有技术的典型代表,采用jsm-6510lv型扫描电子显微镜(sem)观察两个样品的结构和形貌,测试结果分别为附图1和附图2所示。
100.采用美国康塔比表面积及孔径分析仪依次测试实施例1-4及对比例1-2所得碳气凝胶样品的比表面积和孔容积,测试结果如表1所示。
101.2、电极性能测试
102.将实施例1-4及对比例1-2所得碳气凝胶球磨至d50为5-6μm,按碳气凝胶:乙炔黑:sbr:cmc=90:4:3:3的质量比分别配制成粘度为3000
±
300cp的浆料并依次双面涂布于22μm腐蚀铝箔上,经干燥、辊压后得到厚度为220μm的碳气凝胶电极。分别取样测试各个碳气凝胶电极样品的压实密度,测试结果如表1所示。
103.3、φ22
×
45焊针式超级电容器产品性能测试
104.将厚度为220μm的碳气凝胶电极分别用切刀分切成宽35mm,正极长度570mm,负极长度525mm。采用日本nkk公司生产的超级电容器专用tf4035型隔膜与分切好的正负极一起卷绕成电芯。各取10个电芯置于1m四乙基铵四氟硼酸在乙腈中的溶液中真空浸渍至饱和吸液状态,将浸渍好的电芯装入壳内,封口,得到φ22
×
45焊针式超级电容器单体,按1a充放电测试各种单体的初始容量及内阻,计算出对应单体的体积能量密度和功率密度,测试及计算结果如表2所示。
105.表1碳气凝胶比表面积、孔容积及对应的电极压实密度
[0106][0107][0108]
表2 φ22
×
45焊针式超级电容器的电性能
[0109][0110]
对比例1和2均是现有技术中制备碳气凝胶的方法,由表1和表2可知,本技术提供的制备方法得到的碳气凝胶比表面积和孔容积均优于对比例1和2,且本技术的碳气凝胶制备成超级电容器电极后,电极压实密度高,应用于超级电容器后,所得超级电容器兼具高体积能量密度及高功率密度特性。
[0111]
由附图1和2比较可知,本技术提供的碳气凝胶由纳米级细小颗粒堆积而成,颗粒与颗粒之间形成三维连通的整体骨架结构,内部孔隙相对均匀,颗粒的堆积较为致密,有利于提高碳气凝胶的孔容积及超级电容器用电极压实密度。添加表面活性剂p123后常压干燥
制备的碳气凝胶堆积颗粒约2μm,其骨架结构相比本技术的碳气凝胶明显偏粗,颗粒和颗粒之间的孔隙大小不均,颗粒的堆积也较为松散,导致对比例2中碳气凝胶制备成的超级电容器电极压实密度偏低。
[0112]
由表1的测试结果可知,本技术提供的碳气凝胶材料比表面积高、孔容积大,对应的电极压实密度比现有技术制备的碳气凝胶电极压实密度高37%以上。根据超级电容器电性能测试结果,用本发明碳气凝胶制造的φ22
×
45焊针式超级电容器单体容量高、内阻低,能量密度及功率密度相比基于现有技术的碳气凝胶制造的超级电容器产品均有显著提升。此外,由于本发明制备方法简单、生产效率高,非常有利于产业化企业推广使用。
[0113]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1