一种放射性金属卤化物废物的脱卤及玻璃固化方法
1.本发明涉及放射性废物处理技术领域,尤其涉及一种放射性金属卤化物废物的脱卤及玻璃固化方法。
背景技术:
2.核能是一种安全、经济、高效的清洁能源,也是我国实现碳达峰、碳中和目标的主要能源之一。采取核燃料闭式循环的方式,回收并充分利用乏燃料中的铀和钚,即乏燃料后处理,对核能的可持续发展具有重要意义。其中,乏燃料干法后处理因具有耐辐照、低临界风险、可处理对象广、放射性废物少等优点,已成为未来先进燃料循环最有前景的技术之一。
3.在乏燃料干法后处理过程中,常用卤化物作为稀释剂,因此会产生含有金属卤化物的熔盐废物。例如,熔盐电解法采用金属乏燃料作为阳极,氯化物熔盐作为电解质,根据氧化还原电位差,实现锕系元素和裂片元素的分离,并将铀和钚沉积在惰性阴极上。但随裂片元素在熔盐中的富集,锕系元素与裂片元素的分离效率也随之降低,并影响阴极回收产品的质量,此时熔盐需卸载,并需按照放射性废物进行处理与处置。而熔盐堆是以熔融氟化物混合盐作为燃料载体和冷却剂,其乏燃料的干法后处理也通常采用氟化物作为氧化剂和吸附剂,经氟化挥发、减压蒸馏、熔盐萃取等工艺将铀、钍、熔盐载体、裂变核素分离,因此会产生各种类型的含氟放射性废物。
4.鉴于直接和放射性乏燃料接触,这些包含了大量卤素的放射性废物(卤素含量一般大于40wt%)被归为高放废物。对于高放废物的处理,目前国际上比较成熟的方法是利用硼硅酸盐玻璃基材将其固化。然而,卤素在硼硅酸盐玻璃中的低溶解度(一般小于1wt%),限制了玻璃固化体对废物的包容量。此外,高温熔制过程中,卤化物的挥发易造成核素的迁移。因此,现有的玻璃固化工艺已不适于处理放射性金属卤化物废物。
5.针对放射性金属卤化物废物中卤素含量高的特点,目前国际上提出两种处理途径。一种是采用对卤素溶解度大的材料作为固化基材,如美国阿贡国家实验室开发的玻璃粘结方钠石玻璃陶瓷固化体,其处理能力已达300~400kg/批(bateman k.j,morrisona m.c,rappleye d.s,simpson m.f,frank s.m,scale up of ceramic waste forms for electrorefiner salts produced during spent fuel treatment[j],journal of nuclear fuel cycle and waste technology,2015,13:55),但此工艺流程复杂,废物包容量低(8~14wt%)。另一种则是先采用脱卤剂对废物脱卤,再进行固化处理。如韩国原子能研究院采用溶胶-凝胶法制备出的sap(二氧化硅-氧化铝-磷酸盐)复合物作为脱氯剂,在650℃下实现氯化物熔盐废物的高效脱氯(park h.s,kim i.t,cho y.z,eun h.c,lee h.s,stabilization/solidification of radioactive salt waste by using xsio
2-yal2o
3-zp2o5(sap)material at molten salt state[j],environmental science&technology,2008,42:9357),但脱氯后的sap复合物与固化基材存在相容性差的问题。此外,美国太平洋西北国家实验室的研究人员发现nh4h2po4在600℃也能够有效去除氯化物中的氯,但脱氯后
的废物含有大量磷酸盐,需利用磷酸盐玻璃基材对其固化(riley b.j,peterson j.a,vienna j.d,ebert w.l,frank s.m,dehalogenation of electrochemical processing salt simulants with ammonium phosphates and immobilization of salt cations in an iron phosphate glass waste form[j],journal of nuclear materials,2020,529:151949),而磷酸盐玻璃对熔炉耐火材料及金属电极腐蚀极大,很难实现工业应用。
[0006]
上述两种卤化物废物的处理方法各有优缺点,如玻璃粘结方钠石固化工艺虽能包容较多卤素,但工艺流程复杂;脱卤再固化工艺虽可将废物中的卤素先去除,但脱卤后残留的新物质,为后续的固化体开发提出新挑战,且当前脱卤温度高(大于600℃),废物中放射性易挥发核素有高温迁移的风险。因此,有必要开发一种方法可靠、工艺简单的放射性金属卤化物废物安全处理方法。
技术实现要素:
[0007]
有鉴于此,本发明提供一种放射性金属卤化物废物的脱卤及玻璃固化方法,用以解决现有技术中放射性金属卤化物废物处理过程中工艺复杂、脱卤温度高、脱卤后残留新物质与固化基材相容性差的问题。
[0008]
本发明的第一方面提供一种放射性金属卤化物废物的脱卤方法,包括以下步骤:
[0009]
将放射性金属卤化物废物与草酸混合,经热处理使放射性金属卤化物废物中的卤素脱除。
[0010]
本发明的第二方面提供一种玻璃固化方法,包括如下步骤:
[0011]
通过玻璃固化基材对经本发明第一方面提供的放射性金属卤化物废物的脱卤方法处理的脱卤废物进行固化形成玻璃固化体。
[0012]
本发明的第三方面提供一种玻璃固化体,该玻璃固化体通过本发明第二方面提供的玻璃固化方法得到。
[0013]
与现有技术相比,本发明的有益效果为:
[0014]
(1)本发明提供的脱卤方法脱卤温度低、效率高,不但节能、易操控,而且降低了易挥发核素的迁移风险;
[0015]
(2)脱卤处理后无残留新物质,脱卤废物可采用玻璃固化基材固化处理,废物包容量大,且固化体的化学稳定性满足高放玻璃固化体处置要求;
[0016]
(3)本发明提出先使用草酸脱卤,再采用玻璃固化的方法,使现有成熟的玻璃固化工艺处理放射性金属卤化物废物成为可能,工艺简单、实用性强,具有良好的应用前景和社会价值。
附图说明
[0017]
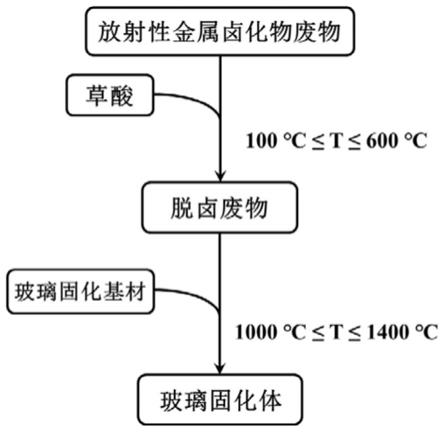
图1是本发明放射性金属卤化物废物的脱卤及玻璃固化方法一实施方式的工艺流程图;
[0018]
图2是本发明实施例1中草酸(h2c2o4)与氯原子的加入比例对脱氯效率的影响结果;
[0019]
图3是本发明实施例1中热处理温度对脱氯效率的影响结果;
[0020]
图4是本发明实施例1保温时间对脱氯效率的影响结果;
[0021]
图5是本发明实施例1制备的玻璃固化体的xrd图;
[0022]
图6是本发明实施例2制备的玻璃固化体的xrd图。
具体实施方式
[0023]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
[0024]
请参阅图1,本发明的第一方面提供一种放射性金属卤化物废物的脱卤方法,包括以下步骤:将放射性金属卤化物废物与草酸混合,经热处理使放射性金属卤化物废物中的卤素脱除。
[0025]
本发明中,热处理温度为100~600℃。进一步为250~500℃,在该温度范围内,脱卤效率可以达到90%以上。更进一步为280~400℃,在该温度范围内,较低的热处理温度能够达到极高的脱卤效率,且降低了易挥发核素的迁移风险。
[0026]
进一步地,热处理时间为20~1000min,进一步为60~600min,更进一步为90~300min。
[0027]
在本发明的一些具体实施方式中,将放射性金属卤化物废物与草酸的混合物置于预先升温至热处理温度的环境中进行热处理。该过程中,热处理的时间优选为30~500min,更优选为60~300min。
[0028]
在本发明的一些具体实施方式中,将放射性金属卤化物废物与草酸的混合物以1~20℃/min的速率随炉升温至热处理温度。该过程中,热处理时间包括升温时间和保温时间。更进一步地,以1~10℃/min的速率随炉升温至热处理温度,保温0~180min。
[0029]
在本发明的一些更具体实施方式中,热处理过程为:以5℃/min的速率随炉升温至300℃,保温0~120min。
[0030]
本发明中,放射性金属卤化物废物和草酸均为固体粉末状,更有利于将二者混合均匀,提高脱卤效果。进一步的,草酸和放射性金属卤化物废物的平均粒径小于100目。
[0031]
在本发明的一些具体实施方式中,热处理前,将放射性金属卤化物废物与草酸固体混合破碎至平均粒径小于100目。
[0032]
本发明中,脱卤过程中,草酸与卤素的摩尔比>0.5。进一步的,草酸与卤素的摩尔比>0.8。更进一步的,草酸与卤素的摩尔比>1。更进一步的,草酸与卤素的摩尔比为1.2~3,在该摩尔比范围内,脱卤效率可达90%以上。更进一步的,草酸与卤素的摩尔比为1.5~2.5,在该摩尔比范围内,脱卤效率可达90%以上,且减少了草酸的用量。
[0033]
在本发明的一些优选实施方式中,草酸与卤素的摩尔比为2。
[0034]
本发明中,放射性金属卤化物废物包括乏燃料干法后处理过程中产生的氯化物熔盐废物、氟化物熔盐废物中的至少一种。进一步地,氯化物熔盐废物包括:碱金属氯化物、碱土金属氯化物、稀土金属氯化物中的至少一种;氟化物熔盐废物包括:碱金属氟化物、碱土金属氟化物、稀土金属氟化物中的至少一种。更进一步的,氯化物熔盐废物包括:licl、kcl、nacl、cscl、srcl2、稀土金属氯化物;氟化物熔盐废物包括:lif、naf、kf、csf、mgf2、srf2、稀土金属氟化物。
[0035]
本发明的第二方面提供一种玻璃固化方法,包括以下步骤:
[0036]
通过玻璃固化基材对经本发明第一方面提供的放射性金属卤化物废物的脱卤方法处理的脱卤废物进行固化形成玻璃固化体。
[0037]
本发明中,玻璃固化基材为硼硅酸盐玻璃。进一步的,玻璃固化基材的化学组成包括:63~70wt%的sio2、17~22wt%的b2o3、6~8wt%的al2o3、5~10wt%的cao。
[0038]
本发明中,按氧化物质量百分比计,玻璃固化基材对放射性废物包容量为15~35%,进一步为20~35%,更进一步为25~35%。
[0039]
本发明中,通过玻璃固化基材对经本发明第一方面提供的放射性金属卤化物废物的脱卤方法处理的放射性废物进行固化形成玻璃固化体的步骤具体为:将脱卤废物与玻璃固化基材混合后,经加热熔融、冷却形成玻璃固化体。
[0040]
本发明中,加热熔融的温度为1000~1400℃,进一步为1100~1200℃;时间为1~6h,进一步为1~3h。
[0041]
在本发明的一些具体实施方式中,加热熔融的温度为1200℃,时间为1~2h。
[0042]
本发明的第三方面提供一种玻璃固化体,该玻璃固化体通过本发明第二方面提供的玻璃固化方法得到。
[0043]
实施例1
[0044]
本实施例选用非放射性氯化物模拟熔盐电解法乏燃料干法后处理产生的电解精炼废盐成分如表1所列。按照不同比例称取草酸和氯化物熔盐废物共20g,充分混合后置于100ml刚玉坩埚内,以5℃/min的速率随炉升温至100~600℃,保温0~120min,随后将坩埚取出,自然冷却至室温得到脱氯废物。脱氯效率(chlorine removal efficiency,cre)按照下列公式进行计算,其中m1和m2分别为原始废物和脱氯废物中cl的质量。
[0045][0046]
表1氯化物熔盐废物成分(wt%)
[0047][0048]
不同草酸与氯的摩尔比、热处理温度和保温时间对脱氯效率的影响分别见图2~4。其中,图2的热处理温度为300℃,保温60min;图3中草酸与氯的摩尔比为2,保温0min;图4中草酸与氯的摩尔比为2,热处理温度为300℃。由图2~4可知,最佳脱氯条件为:草酸与氯的摩尔比为2,随炉升温至300℃,保温60min,脱氯效率可达99%。对最佳脱氯条件下得到的脱氯废物进行玻璃固化,按表2设计的玻璃固化配方组成(废物包容量为35wt%),称取20g
混合料,充分混合后置于50ml刚玉坩埚,在1200℃马弗炉中保温1h后将玻璃液倒入预热的铜板模具内,冷却得到玻璃固化体。
[0049]
表2实施例1脱氯废物及玻璃固化基材在所设计玻璃固化配方中的占比(wt%)
[0050][0051]
本实施例制得固化体的xrd衍射图谱(图5)为典型的非晶态衍射峰,证明了制备的样品为玻璃态,阿基米德排水法测得其密度为2.58g/cm3。利用pct-7评估玻璃固化体样品的化学稳定性。表3为本实施例主要元素的pct-7浸出结果,各元素浸出值均低于2g/m2,满足高放玻璃固化体化学稳定性国际要求。
[0052]
表3实施例1玻璃固化体主要元素pct-7浸出结果
[0053]
[0054][0055]
实施例2
[0056]
本实施例选用非放射性氟化物模拟熔盐堆乏燃料干法后处理产生的含氟废物,成分如表4所列。按照草酸与氟的摩尔比为2,称取草酸和氟化物熔盐废物共20g,充分混合后置于100ml刚玉坩埚,以5℃/min的速率随炉升温至300℃保温60min,随后将坩埚取出,自然冷却至室温得到脱氟废物。脱氟效率(fluorin removal efficiency,fre)按照下列公式进行计算,其中m3和m4分别为原始废物和脱氟废物中f的质量。
[0057][0058]
表4氟化物熔盐废物成分(wt%)
[0059][0060]
通过上述脱氟方法,氟化物熔盐废物中氟的去除效率可达91%。对脱氟废物进行玻璃固化,按表5设计的玻璃固化配方组成(废物包容量为25wt%),称取20g混合料,充分混合后置于50ml刚玉坩埚,在1200℃马弗炉中保温1h后将玻璃液倒入预热的铜板模具内,冷却得到玻璃固化体。
[0061]
表5实例2脱氟废物及玻璃固化基材在所设计玻璃固化配方中的占比(wt%)
[0062][0063]
本实施例制得固化体的xrd衍射图谱(图6)为典型的非晶态衍射峰,证明了制备的样品为玻璃态,阿基米德排水法测得其密度为2.48g/cm3。利用pct-7评估玻璃固化体样品的化学稳定性。表6为本实施例主要元素的pct-7浸出结果,各元素浸出值均低于2g/m2,满足高放玻璃固化体化学稳定性国际要求。
[0064]
表6实施例2玻璃固化体主要元素pct-7浸出结果
[0065][0066][0067]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1