金属相变化合物及其制备方法与流程
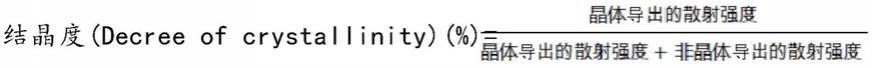
1.本发明提供一种金属相变化合物及其制备方法。
背景技术:
2.物质具有固有的晶界,其晶体习性(crystal habit)根据外部条件而变化。晶面的生长会受到外部条件(如过饱和度、温度、溶剂种类、溶液ph值、杂质和搅拌速度等)的影响。也就是说,即使是相同成分的物质,在外部条件不同的情况下,也可能出现结晶成不同形状的现象,根据物质的内部结构,大致可分为晶型和无定形。晶型是基本结构单元晶胞(unit cell)中构成成分呈一定排列,使得晶体在热力学上稳定。因此,晶型结构中包含的活性成分可保持稳定,但是不易溶解,在晶型结构的情况下,可能难以释放活性成分(例如药物)。另一方面,在无定形结构的情况下,由于内部结构无序排列,具有热力学上较为不稳定的特性,该特性如下。
3.i)与晶型相比,无定形结构体中的药物比较易于溶解和吸收,因为溶解时不需要克服晶型中出现的晶格能。因此,无定形具有相对高的平衡浓度(溶解度),故生物利用度(bioavailability:ba)较为优异。
4.ii)无定形虽不如晶型稳定,但由于是热力学上不稳定的物质,在放置不理的情况下,有时会自然结晶。
5.iii)与晶型相比,无定形结构体中的药物比较易于溶解和吸收,因为溶解时不需要克服晶体的晶格能。因此,与晶型结构相比,无定形结构体中包含的药物通常显示更高的溶解度和更快的溶出率。
6.也就是说,对于结晶性固体,元素之间的排列是能级最低的排列,而无定形固体的元素排列会保持具有高能级的不稳定状态。因此,在相同物质的情况下,结晶性固体比无定形固体在化学上更稳定,并且具有低溶解度和高密度,因此具有要保持热力学上稳定的晶体形态的特征。
7.金属化合物或无机化合物(如氧化锌或氢氧化锌)作为结晶性很高的溶剂化物(solvate)或水合物(hydrate)类结构体,制备时温度、溶剂、过饱和度、核生成(seed becoming)/生长速度(seed growth speed)、结晶机理、结晶时间等是生成晶体的主要因素,这些各种因素归因于晶体生成。也就是说,常规金属氢氧化物具有非常高的结晶趋势,因此难以在单体中形成高无定形性。
8.目前也有与金属氢氧化物相关的诸多技术,如韩国授权专利第10-1738545号所述的与结晶性高的金属氢氧化物及利用它的纳米复合物相关的技术,但是找不到无定形性高的金属氢氧化物。
9.因此,为了解决上述的问题提出了本发明。
技术实现要素:
10.技术问题
11.本发明涉及一种通过提高金属化合物中的无定形性而同时具有无定形性和结晶性的金属相变化合物(metal phase transformation compound)。具体地,本发明的金属相变化合物是衍生自dds(药物传递系统)概念的药物载体的一种,其稳定地保持不稳定活性成分,具有在水溶液状态的一定条件下使活性成分溶出的特征。
12.在本发明中,相变是指多晶型物(polymorph),并且是指金属化合物中同时具有无定形性和结晶性的无定形-晶质(co amorphous-crystalline)金属化合物。本发明的金属相变化合物在溶解度、药物含量等方面具有不同于结晶性化合物的物性,并且显示出不同于现有晶型的特征。多晶型晶体的物理化学特性之差归因于晶体结构中相邻分子的取向以及分子之间的相互作用之差,物质中构成元素的排列在固态的热力学能稳定性上也有很多差异。
13.因此,本发明旨在提供一种金属化合物的一个单体中同时具有无定形性和结晶性特征的金属化合物。更具体地,本发明旨在提供一种金属相变化合物,该金属相变化合物包含通过无定形性的最大化结晶相处于无定形和晶型混合并存的状态的无定形-晶质。
14.技术方案
15.本发明提供一种金属相变化合物,其包含肽,并且包含60体积%以上的无定形。
16.发明效果
17.本发明的金属相变化合物以高比例具有单体中的无定形性,从而可以使金属相变化合物中有效成分(即肽)的含有率最大化。
18.此外,本发明的金属相变化合物同时具有结晶性和无定形性,并且以高比例具有无定形性,从而可以稳定地承载活性成分,并且具有优异的生物利用度、溶解度、溶出率。
19.另外,本发明能够提供可有效地形成具有无定形性的金属相变化合物的制备发明。
附图说明
20.图1是本发明的金属相变化合物形成方法的方案,具体示出通过成核(nucleation)所生成的晶种(seed)根据结晶相(crystalline phase)的生长(growth)和扩散相变(diffussional phase transformation)由粒子中原子的移动生成和生长出共格界面(coherent interface)部分的过程。因此,随着短程有序相(short-range order phase)与周期有序相(periodic order phase)一起形成和递增,初始晶种(initiation seed)堆叠(stacking)而形成粒子(particle)。
21.图2是现有含活性成分的金属氢氧化物复合物制备方法的现有方法(preexistence method)以及本发明的金属相变化合物制备方法的新方法(new method)的示意图。
22.图3是与晶型比相关的结晶度的图。
23.图4是实施例1至5的xrd图与参考例的不含肽的结晶性zbs的xrd图的比较图。
24.图5是实施例6至8的xrd图与参考例的不含肽的结晶性zbs的xrd图的比较图。
25.图6是实施例1的xrd图。
26.图7是实施例2的xrd图。
27.图8是实施例3的xrd图。
28.图9是实施例4的xrd图。
29.图10是实施例5的xrd图。
30.图11是实施例6的xrd图。
31.图12是实施例7的xrd图。
32.图13是实施例8的xrd图。
33.图14是比较例的xrd图。
34.图15是示出实施例3的sem-edx结果的图。
35.图16是关于实施例3(peptibrid500ppm)、实施例3(peptibrid1000ppm)、pal-ghk(500ppm)和pal-ghk(1000ppm)的穿透率的图。
36.图17是关于实施例3(500ppm)的粒子大小的图。
37.图18是关于实施例3(1000ppm)的粒子大小的图。
38.图19是关于pal-ghk(500ppm)的粒子大小的图。
39.图20是关于pal-ghk(1000ppm)的粒子大小的图。
40.图21是将实施例3的化合物放大1000倍的fe-sem扫描电子显微镜图片。
41.图22是将实施例3的化合物放大10000倍的fe-sem扫描电子显微镜图片。
42.图23是将实施例3的化合物放大10000倍的fe-sem扫描电子显微镜图片。
43.图24是将实施例3的化合物放大20000倍的fe-sem扫描电子显微镜图片。
44.图25是将实施例3的化合物放大100000倍的fe-sem扫描电子显微镜图片。
45.图26至29是实施例3的tem透射电子显微镜图片。
具体实施方式
46.本发明通过提高金属化合物中的无定形性,可以使金属化合物中活性成分含量增加以及具有优异的缓释性。通常,金属化合物具有结晶性高的结晶金属化合物的特征。这种结晶金属化合物(crystalline metal compound)是热力学上非常稳定的物质,并具有内部规则性优异的结晶性。在这种结晶性高的金属化合物中,通过物理/化学方法来诱导规则性变形,以引起(多晶型)相变,并且衍生的异晶质部分(无定形,amorphous)与阴离子性物质层叠而一起形成无定形-晶质(co amorphous-crystalline)。对于如此形成的金属盐,由于内部结构改变,具有无定形-晶质结构的金属化合物中包含的活性成分含量高于原结晶性金属化合物所具有的活性成分含量的特征,并且具有根据ph变化活性成分的释放量被调节的特征。
47.具体地,本发明涉及一种金属相变化合物,其特征在于,所述金属相变化合物包含肽,并且包含60体积%以上的无定形。也就是说,本发明是将无定形性引入传统的有限金属化合物结构中,以形成同时具有无定形性和结晶性特征的多晶型物(polymorph)结构的金属化合物。多晶型物结构具有相同的化学组分,但分子形成不同的排列,在不同的范德华力、氢键和分子之间的相互作用等物理力量的作用下晶核(seed)生长,因此具有不同于现有金属化合物的晶体结构。在本发明中,利用金属化合物对混合溶剂的反应性(polarity)和溶解度来改变金属化合物内部结构的对称性和可重复性,由此改变金属化合物中彼此接触的原子之间的距离,人为降低结晶性使金属化合物变成无定形结构,从而提出了具有结晶相转变的无定形-晶质(co amorphous-crystalline)的金属相变化合物。
48.具体地,所述金属相变化合物可包含1至40体积%的晶型和99至60体积%的无定形,优选包含1至35体积%的晶型、99至65体积%的无定形,进一步优选包含1至30体积%的晶型、99至70体积%的无定形,更优选包含1至25体积%的晶型、99至75体积%的无定形,更进一步优选包含1至15体积%的晶型、99至85体积%的无定形。
49.当金属化合物包含上述范围的无定形时,与热力学上稳定的原子排列而导致溶解度降低的纯结晶性金属化合物相比,含活性成分(如肽)的无定形-晶质(co amorphous-crystalline)的金属相变化合物可以根据ph调节粒子大小,并且可以提高所含活性成分的释放速度。再者,将所述金属相变化合物应用于化妆品制剂后涂抹在皮肤时,可以期待通过皮肤所具有的ph范围(ph4.5~7)来提高完整地传递用户所需成分的能力。
50.本发明的金属相变化合物具体可为含肽的金属氧化物或金属氢氧化物复合物,对此没有限制,优选可为由下述化学式1表示的化合物。
51.[化学式1]
[0052]
{m
2+
(oh)y(o)z}a
(2-x
′
)
·
nh2o
[0053]
在所述化学式1中,
[0054]m2+
为mg
2+
、ca
2+
、co
2+
、cu
2+
、ni
2+
或zn
2+
,
[0055]
a为生理活性物质,
[0056]
x
′
为1以上且小于2的数,
[0057]
y为0以上且2以下的数,
[0058]
z为0以上且2以下的数,
[0059]
y+z不大于2,
[0060]
y和z不同时为0,
[0061]
n为0以上且10以下的数。
[0062]
在所述化学式1中,a的生理活性物质更优选可为肽。
[0063]
本发明中包含的肽可以是pi值为2至12的肽。上述的肽与pka密切相关,在ph为5至10以下的情况下,与金属离子形成配位共价键(coordinate covalent bond),氢键(hydrogen bonding)、羧酸盐键(carboxylate bonding)等键合活性基团(bonding active group)的活化位点(activation site)具有通过pi可提高无定形性的效果。具体地,所述肽可为选自二肽-1(yr,(pi=,以下省略)9.95)、二肽-2(vw,5.98)、二肽-4(fw,5.98)、二肽-6(kv,9.07)、二肽-7(kt,9.07)、二肽-14(at,5.98)、gh二肽(gh,7.37)、乙酰基二肽-1(yr,9.95)、乙酰基二肽-1鲸蜡酯(yr,9.95)、烟酰基二肽-2(vw,5.98)、cp二肽(cp,5.21)、vge二肽(ve,3.64)、cge二肽(ce,3.64)、ege二肽(ee,3.46)、tge二肽(te,3.64)、lge二肽(le,3.64)、eq二肽(eq,3.64)、gr二肽(gr,10.47)、hg二肽(hg,7.37)、pe二肽(pe,3.64)、de二肽(de,3.29)、hq二肽(hq,7.37)、rs二肽(rs,10.47)、hp二肽(hp,7.37)、肌肽(ah,7.37)、三肽-1(ghk,9.07)、三肽-3(ghr,10.47)、三肽-4(lgd,3.37)、三肽-5(kvk,9.37)、三肽-6(gxp,5.98)、三肽-8(hfr,10.47)、三肽-10(kdi,6.34)、rgd肽(rgd,6.5)、ahk肽(ahk,9.07)、三肽-29(gpx,5.98)、三肽-54(fty,5.98)、生物素三肽-1(ghk,9.07)、硫代辛酰基三肽-1(thioctoyl tripeptide-1)(ghk,9.07)、三肽(rfk,10.61)、hgg肽(hgg,7.37)、rkr肽(rkrm,11.84)、四肽-1(lptv,5.98)、四肽-2(kdvy,6.34)、四肽-3(kghk,9.37)、四肽-5(ahsh,7.52)、四肽-7(gqpr,10.47)、四肽-9(qdvh,4.78)、四肽-11(ppyl,5.98)、四肽-15
(ypff,5.98)、四肽-21(gekg,6.6)、四肽-26(elps,3.64)、乙酰基四肽-2(kdvy,6.34)、乙酰基四肽-3(kghk,9.37)、乙酰基四肽-5(ahsh,7.52)、乙酰基四肽-9(qdvh,4.78)、乙酰基四肽-11(ppyl,5.98)、乙酰基四肽-15(ypff,5.98)、五肽-3(gprpa,10.47)、五肽-4(kttks,9.37)、五肽-17(klakk,9.54)、五肽-18(yagfl,5.98)、硫代辛酰基五肽-4(thioctoyl pentapeptide-4)(kttks,9.37)、六肽-1(arhlfw,10.47)、六肽-2(fwfkpv,9.07)、六肽-3(eemqrr,6.91)、六肽-4(fghxaf,7.37)、六肽-5(fgvxaf,5.98)、六肽-6(vepipy,6.91)、六肽-9(gpqgpq,5.98)、六肽-11(fvapfp,5.98)、六肽-12(vgvapg,5.98)、乙酰基六肽-3(eemqrr,6.91)、乙酰基六肽-8(eemqrr,6.91)、七肽-6(hwawfk,9.07)、半胱氨酸肽(rfaacaa,8.33)、棕榈酰二肽-6(kv,9.07)、棕榈酰二肽-7(kt,9.07)、壬二酰三肽-1(ghk,9.07)、棕榈酰-三肽-3(ghr,10.47)、棕榈酰三肽-5(kvk,9.37)、棕榈酰三肽-1(ghk,9.07)、棕榈酰三肽-5(kvk,9.37)、棕榈酰三肽(rfk,10.61)、肉豆蔻酰三肽-1(ghk,9.07)、棕榈酰三肽-4(lgd,3.37)、棕榈酰三肽-8(hfr,10.47)、棕榈酰四肽-7(gqpr,10.47)、肉豆蔻酰五肽-17(klakk,9.54)、棕榈酰五肽-4(kttks,9.37)、棕榈酰五肽-17(klakk,9.54)、肉豆蔻酰六肽-12(vgvapg,5.98)和棕榈酰六肽-12(vgvapg,5.98)中的一种以上肽,并且大小可为2mer以上且10mer以下。当使用所述范围的肽时,可以显示出优异的无定形性。
[0064]
本发明的特征是包含在15至25
°
范围内形成的宽范围的x射线衍射图谱。在所述范围内形成的峰可以形成较宽的单一值峰,但是也可以形成所述范围内包含一个以上小峰的图谱。当包含如上所述的峰图谱时,可能意味着无定形性高。对于结晶性高的金属化合物,在特定值下形成范围窄而高的峰,而在宽范围内形成的峰可能意味着一个单体中具有高比例的无定形性。具体地,如图26至29的tem图像所示,本发明是无定形和晶型并存的金属化合物,因此在15至25
°
范围形成的显示无定形性的宽峰范围内可包含一个以上的显示晶型的小峰。也就是说,除了无定形的峰之外,还可以出现无定形峰中包含显示结晶性的峰。
[0065]
另外,对于本发明的金属相变化合物,粉末-x射线衍射图谱包含衍射角(2θ)=19
±6°
、33
±5°
和59
±5°
的峰值,在15至25
°
范围形成的峰中可包含一个以上的峰。
[0066]
另外,对于本发明的金属相变化合物,1单体中肽可包含20重量%以上,进一步优选可包含30重量%以上,更优选可包含40重量%以上,更进一步优选可包含50重量%以上。
[0067]
另外,对于本发明的金属相变化合物,1单体中可包含20至80重量%的肽和10至35重量%的金属,进一步优选可包含30至80重量%的肽和10至35重量%的金属,更优选可包含40%至80重量%的肽和10至35重量%的金属。
[0068]
因此,本发明可以提供包含上述的金属相变化合物的皮肤外用组合物。所述皮肤外用组合物预期可以用作通过改善功能性肽的皮肤传递效率具有改善的功能性的化妆品、准药物和卫生产品的原料。另外,所述皮肤外用组合物可以制成各种制剂,例如乳膏、凝胶、软膏、润肤液、精华液等。
[0069]
对于本发明的金属相变化合物,其制备方法不受限制,只要是现有的金属化合物的制备方法即可。当制备方法包含将金属溶液、肽溶液和氢氧化物溶液各自分开制备的步骤和将所述分开制备的溶液进行混合的步骤时,可以提高金属化合物中的无定形性,从这一点上看优选该制备方法。
[0070]
另外,在所述制备方法中,将整个制备方法所使用的总溶液和溶剂进行换算时,所使用的总溶液和溶剂中乙醇与水的比例是以体积比计,乙醇∶水为10∶90至90∶10的范围。尤
其,本发明中使用的乙醇更优选可以是纯度为95%以上的乙醇。当溶液和溶剂的水与乙醇的比例满足所述范围时,可以提高金属化合物中的无定形性,从这一点上看优选所述范围。
[0071]
《金属相变化合物的制备方法》
[0072]
在本发明中,当合成金属相变化合物时,所有工艺都是在常温(20℃)和氮气环境下进行。另外,使用了95%乙醇和三次蒸馏水,并且使用了纯度为95%以上(水分含量为5%以下)的pep(肽)。naoh水溶液是3.2m,用三次蒸馏水来制备。按照合成-洗涤-干燥的顺序如下实施。向罐体1加入肽(pep)相当于0.4当量(equiv.)~0.13当量的重量,然后将三次蒸馏水和乙醇(95%《)相对于乙醇保持30%以上的共溶剂(co-solvent)加入罐体1中进行搅拌。向罐体2加入氧化锌(zno)相当于1当量的重量,然后将辅助罐体1的浓盐酸(conc.hcl)溶液缓慢滴加到主罐体并滴定成ph为0.5~1,在氮气环境下以700rpm搅拌30分钟使其溶解。
[0073]
然后,准备罐体3的3.2m的naoh溶液,向主罐体一并加入罐体1、2、3的溶液后,滴定成主罐体的ph保持6.5~7.5,主罐体中搅拌3小时,并引起沉淀反应,ph保持为6.5~7.5。
[0074]
接下来是沉淀物的洗涤过程,为了去除所述溶液中未反应的盐和离子性物质,使用离心机去除杂质。离心分离是以9000rpm进行四次,每次为5分钟,以将沉淀物与溶液分离,其顺序如下。将合成的沉淀物溶液以9000rpm离心分离5分钟。将溶液与沉淀物分离后,再将沉淀物与乙醇和三次蒸馏水以1∶1的比例均匀地稀释,然后以9000rpm进行搅拌5分钟。该过程重复四次。最后离心分离洗涤只用蒸馏水稀释,然后以9000rpm进行离心分离10分钟,从而获得沉淀物。
[0075]
实施例:实施例1至8的金属相变化合物
[0076]
实施例1的合成
[0077]
作为实验基本条件,所有工艺都是在常温(20℃)和氮气环境下进行。另外,使用了95%乙醇和三次蒸馏水,并且使用了纯度为95%以上(水分含量为5%以下)的pep。naoh水溶液是3.2m,用三次蒸馏水来制备。
[0078]
按照合成-洗涤-干燥的顺序如下实施。向罐体1加入棕榈酰-ghk(2.845g)和乙醇(30ml)后进行搅拌。向罐体2加入zno(1g)、三次蒸馏水(5ml)、乙醇(15ml)后,将辅助罐体1的浓盐酸溶液滴加到主罐体并滴定成ph为1(0.5《)以下,在氮气环境下以500rpm搅拌30分钟使其溶解。
[0079]
然后,准备罐体3的3.2m的naoh溶液,向主罐体一并加入罐体1、2、3的溶液后,滴定成主罐体的ph保持6.5~7.5,主罐体中以700rpm搅拌3小时,并引起沉淀反应,ph保持为6.5~7.5(相对于乙醇,总体溶剂加入量保持在30%以上)。
[0080]
接下来是沉淀物的洗涤过程,为了去除所述溶液中未反应的盐和离子性物质,使用离心机去除杂质。离心分离是以9000rpm进行四次,每次为5分钟,以将沉淀物与溶液分离,其顺序如下。将合成的沉淀物溶液以9000rpm离心分离5分钟。将溶液与沉淀物分离后,再将沉淀物与乙醇和三次蒸馏水以1∶1的比例均匀地稀释,然后以9000rpm进行搅拌5分钟。该过程重复四次。最后离心分离洗涤只用蒸馏水稀释,然后以9000rpm进行离心分离10分钟,从而获得沉淀物。用于具体合成的化合物和合成结果示于下表1中。
[0081]
晶型比是通过如下式来确定,图3中示出与晶型相关的图。
[0082]
[式1]
[0083][0084]
实施例2至5
[0085]
通过与所述实施例1的合成方法相同的方法按照表1所示的内容合成了实施例2至5的金属相变化合物。合成结果也示于下表1中。
[0086]
【表1】
[0087]
[0088][0089]
(*pep1:棕榈酰-ghk)
[0090]
对于现有的晶型金属氢氧化物复合物,棕榈酰-ghk最多可以含有40%。然而,从所述实施例1至5来看,金属相变化合物的肽含量为60%以上,可以确认活性成分含量特征得到了明显改善。另外,为此,通过实验过程中调节乙醇的比例来人为诱导无定形-晶质(co amorphous-crystalline),其结果可以通过xrd图谱(pattern)来确认。
[0091]
实施例6至8
[0092]
除了含量和肽的种类等表2所示的内容之外,通过与所述实施例1的合成方法相同
的方法来合成了实施例6至8的金属相变化合物。合成结果也示于下表2中。
[0093]
【表2】
[0094][0095]
(*pep2:棕榈酰-五肽4)
[0096]
对于现有的晶型金属氢氧化物复合物,棕榈酰-五肽4最多可以含有20%。然而,从所述实施例6至8来看,金属相变化合物的肽含量为60%以上,可以确认活性成分含量特征得到了明显改善。另外,为此,通过实验过程中调节乙醇的比例来人为诱导无定形-晶质(co amorphous-crystalline),其结果可以通过xrd图谱(pattern)来确认。
[0097]
参照例:层状氢氧化锌(zinc layered hydroxide)的制备方法
[0098]
将zn(no3)2·
6h2o(5g)溶解于去除碳酸根(co
32-)的三次蒸馏水中,然后使用0.2m的naoh将ph滴定成6~7,从而获得了碱性锌盐(zinc basic salt)沉淀物。将所述滴定的溶液用离心机进行分离,通过洗涤过程去除未反应的盐,从而制成白色粉末形式2.6g(收率为70%)。
[0099]
比较例:含肽结晶性金属氢氧化物的制备方法
[0100]
作为实验基本条件,所有工艺都是在常温(20℃)和氮气环境下进行。另外,使用了95%乙醇和三次蒸馏水,并且使用了纯度为95%以上(水分含量为5%以下)的肽。naoh水溶液是3.2m,用三次蒸馏水来制备。
[0101]
本发明中使用的肽是棕榈酰ghk。
[0102]
实验是按照合成-洗涤-干燥的顺序如下实施。
[0103]
向主罐体加入zn(no3)26h2o(3g)和乙醇(25ml)、三次蒸馏水(25ml)后,以700rpm搅拌30分钟使其溶解。然后,在辅助罐体1中溶解肽(0.58g)和乙醇(20ml)、三次蒸馏水(10ml)后加入主罐体(相对于zn加入量,总体溶剂加入量为25体积)以300rpm搅拌5分钟使其溶解。保持主罐体的搅拌速度,并将辅助罐体2的3.2m的naoh溶液加入主罐体,然后滴定成主罐体的ph保持6.5~7.5,主罐体中搅拌3小时,并引起沉淀反应,ph保持为6.5~7.5。
[0104]
接下来是沉淀物的洗涤过程,为了去除所述溶液中未反应的盐和离子性物质,使用离心机去除杂质。离心分离是以8000rpm进行三次,每次为5分钟,以将沉淀物与溶液分离,其顺序如下。将合成的沉淀物溶液以8000rpm离心分离5分钟。将溶液与沉淀物分离后,再将沉淀物与乙醇和三次蒸馏水以1∶1的体积比均匀地稀释,然后以8000rpm进行搅拌5分钟。该过程重复三次。最后离心分离洗涤只用乙醇稀释,然后以9000rpm进行离心分离10分钟,从而获得沉淀物。
[0105]
所述比较例的1单体中包含的肽的含量为20%,图14中示出xrd图。
[0106]
实验例1:金属化合物的xrd峰的测定
[0107]
仪器:粉末-x射线衍射(pxrd)
[0108]
x射线衍射仪(d/maxprint 2200-ultima,理学,日本)
[0109]
cu-kα辐射
[0110]
管电压(tube voltage)为40kv,电流为30ma
[0111]
用于测定的x射线衍射仪采用了理科(日本)公司的d/maxprint 2200-ultima。用于产生x射线的阴极使用了cu金属,用于测定的是kα射线测定范围为2θ=3~70
°
,扫描速度(scanning speed)为0.02
°
/0.2秒,发散狭缝(divergence slit)、散射狭缝(scattering slit)和接收狭缝(receiving slit)分别为0.1、1和1mm。管电压(tube voltage)为40kv,电流为30ma。
[0112]-评价标准
[0113]
对z轴的一维电子密度(one-dimensional(1d)electron density)通过如下方程算出。
[0114]
[方程1]
[0115][0116]
对经由合成获得的粉末通过xrd衍射图谱进行比较分析,基于此的层距是通过布拉格方程(bragg
′
s equation;下述方程2)进行了计算。在位于最前的峰(peak)的情况下,表示合成的金属化合物的层与存在阴离子的层的距离包含在内的层距,可称为主要层距。
[0117]
[方程2]
[0118]
nλ=2dsinθ
[0119]
(λ=x射线的波长,d=晶体的晶格间距,θ=入射角)
[0120]
测定的xrd图谱示于图4至图10中。
[0121]
从图4至图10可知,在15至25
°
范围形成宽峰。也就是说,本发明的金属相变化合物是人为诱导了各单体中的无定形-晶质(co amorphous-crystalline),这一点可以通过所述xrd图谱来确认。
[0122]
具体地,图6的粉末-x射线衍射图谱可包含衍射角(2θ)为7.02
±1°
、8.36
±1°
、19.78
±6°
、20.66
±1°
、33.3
±5°
、47.43
±1°
、59.33
±5°
、69.5
±1°
和76.5
±1°
的峰值,更具体地具有7.02
±1°
、8.36
±1°
、19.78
±1°
、20.66
±1°
、33.3
±1°
、47.43
±1°
、59.33
±1°
、69.5
±1°
和76.5
±1°
的峰,在15-25
°
范围形成宽范围的峰。
[0123]
具体地,图7的粉末-x射线衍射图谱包含衍射角(2θ)为18.86
±1°
、19.81
±6°
、33.3
±5°
、59.12
±5°
和69.37
±1°
的峰值,更具体地具有18.86
±1°
、19.81
±1°
、33.3
±1°
、59.12
±1°
和69.37
±1°
的峰,在15-25
°
范围形成宽范围的峰。
[0124]
具体地,图8的粉末-x射线衍射图谱包含衍射角(2θ)为4.87
±1°
、9.29
±1°
、21.25
±6°
、33.086
±5°
、59.15
±5°
和69.30
±1°
的峰值,更具体地具有4.87
±1°
、9.29
±1°
、21.25
±1°
、33.086
±1°
、59.15
±1°
和69.30
±1°
的峰,在15-25
°
范围形成宽范围的峰。
[0125]
具体地,图9的粉末-x射线衍射图谱包含衍射角(2θ)为8.4
±1°
、19.57
±6°
、33.155
±5°
、59.19
±5°
和69.46
±1°
的峰值,更具体地具有8.4
±1°
、19.57
±1°
、33.155
±1°
、59.19
±1°
和69.46
±1°
的峰,在15-25
°
范围形成宽范围的峰。
[0126]
具体地,图10的粉末-x射线衍射图谱包含衍射角(2θ)为3.97
±1°
、20.43
±6°
、33.196
±5°
和59.21
±5°
的峰值,更具体地具有3.97
±1°
、20.43
±1°
、33.196
±1°
和59.21
±1°
的峰,在15-25
°
范围形成宽范围的峰。
[0127]
实验例2:sem-edx(扫描电子显微镜-能量色散x射线光谱仪)
[0128]
用于sem-edx的仪器使用了场发射扫描电子显微镜(jeol-7610f-plus),是用于纳米结构材料和元件的结构分析和成像的分析装置。
[0129]
1.性能
[0130]
1)分辨率:0.8nm(15kv)
[0131]
1.0nm(1kv gb模式)
[0132]
3.0nm(15kv,pc 5na,wd 8mm)
[0133]
2)放大率:直接x25至x1000000
[0134]
显示x75至3000000
[0135]
3)加速电压:0.01kv至30kv
[0136]
4)探针电流:几pa至≥200na
[0137]
2.电子枪
[0138]
1)类型:浸没式肖特基场发射电子枪
[0139]
2)电子探测器系统:上探测器
[0140]
r型过滤器、内置、下探测器
[0141]
3)eds(能量色散光谱仪):利用x射线的成分定性/定量分析
[0142]
对实施例3的金属相变化合物的sem-edx进行测定,将其结果示于图15和表3中。
[0143]
【表3】
[0144]
元素wt%wt%西格玛原子%c50.630.3667.04n14.000.4515.89o11.280.2011.22zn24.090.225.86
合计100.00 100.00
[0145]
实验例3:金属相变化合物和肽(palghk)的粒子大小以及弱酸性溶剂中浓度差引起的溶解(或分散)确认实验
[0146]
《实验方法》
[0147]
1.粒度(particle size)评价
[0148]
仪器:zetasizer nano zs90
[0149]
分散剂:水
[0150]
分散剂ri:1.330
[0151]
电池:一次性电池
[0152]
运行数:20
[0153]
温度:25℃
[0154]
对于粉末的粒子大小,通过利用电泳光散射的马尔文帕纳科公司(uk)的电位分析仪进行测定,使用nano zs zs90型号测定试样分散于三次蒸馏水后的粒子大小。
[0155]
dls分析结果显示,穿透的肽在500ppm下平均为13.09nm,在1000ppm下为12.18nm,dls分析结果显示,穿透的本发明的含肽金属相变化合物在500ppm下平均为247.6nm,在1000ppm下为256nm。评价结果示于图16至19中。
[0156]
2.分散性评价
[0157]
使用pvdf 0.45μm过滤器
[0158]
搅拌24小时进行hplc测定
[0159]
保持类似于体内温度的温度条件(38℃)
[0160]
溶剂相同,是ph 4.5的缓冲液,以样品原料的肽含量为准是500、1000ppm。
[0161]
以金属化合物中的肽含量(约71%)为准计算浓度。
[0162]
以肽(palghk)中的肽含量(约83%)为准计算浓度。
[0163]
通过所述方法评价分散性的结果示于下表4中。
[0164]
【表4】
[0165][0166]
实验结果显示,肽在弱酸性(ph4.5)下粒子大小减小,直至经过12小时,对于500ppm,过滤器穿透率最高增加到约35%,对于1000ppm,过滤器穿透率最高增加到47%,此后预计会减少。另一方面,本发明的含肽金属相变化合物在弱酸性(ph4.5)下粒子大小减小,直至经过12小时,整体粒子大小减小,对于500ppm,过滤器穿透率最高增加到约56%,对于1000ppm,过滤器穿透率最高增加到66%。在所述dls分析中,穿透的肽与穿透的本发明的
金属相变化合物相比,粒子大小更小,而实际上穿透过滤器的粒子是本发明的金属相变化合物更多,由此预计本发明的金属相变化合物的效率更优异。
[0167]
实验例4:基于fe-sem分析、扫描电子显微镜图片(field emission scanning electron microscope)的评价
[0168]
用扫描电子显微镜拍摄通过实施例3合成的粉末的结果确认,如图21所示形成了大小为几十纳米(nm)至几百微米(μm)的金属相变化合物。以所述图21为准,当进一步放大10倍时,可以确认如图22和图23所示的粒子。从图22和图23也可以确认,本发明的金属相变化合物是小粒子凝聚而形成的。以图21为准,当进一步放大20倍时,如图24所示,当进一步放大100倍时,如图25所示。尤其,从最放大的图25来看,可以确认圆形的小粒子凝聚的形状。这与前面实验的分散性实验相关,虽然金属相变化合物的粒子大小是几十微米以上的大小,但是将对ph的敏感的本复合物在特定ph条件下进行0.45微米(450纳米)过滤处理时,可以确认更多的粒子通过的结果,由此可以容易调节金属相变化合物的粒子大小。
[0169]
实验例5:基于tem分析、透射电子显微镜图片(transmission electron microscope)的评价
[0170]
电子显微镜图片是本发明的实施例3的金属相变化合物的电子显微镜图片,使用了通过产生电子束使其穿透试样来观察和图像化各种物质的内部/晶体结构以及分析化学成分的装置。
[0171]
仪器构成
[0172]-电子枪类型:肖特基场发射枪
[0173]-分辨率:0.23nm(at tem)/0.19nm(at stem)
[0174]-放大倍率:x20至x2.0m(at tem)/x100至x150m(at stem)
[0175]-加速电压:80/120/200kv
[0176]-倾斜角度:
±
30
°
[0177]
用所述电子显微镜测定了通过实施例3合成的金属相变化合物的结构,其结果示于图26至图29中。从图26和图27可以确认,本发明的相变金属化合物是小粒子凝聚而形成了一个大粒子。另外,图27进一步放大了粒子,可以看出小粒子层叠而形成了粒子。再者,从进一步放大粒子大小的图28和图29可以看出,在无定形性状中清晰地观察到阴影,而在有晶质的部分中出现了层状结构或晶格纹路的特征,可以确认本发明的金属相变化合物属于无定形性和晶型并存的化合物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1