一种萜类小分子组装的氧化还原响应光敏药物的制备方法与流程
本发明属于生物医药材料领域,具体涉及一种萜类小分子组装的氧化还原响应光敏药物的制备方法。
背景技术:
天然萜类小分子由于其独特的生理活性如抗癌活性、抗免疫活性和抗菌活性,以及特殊的生物相容性、生物易降解性及生物安全性,在生物医学抗癌方面呈现巨大的前景,将具有抗癌活性的天然萜类小分子用于肿瘤联合治疗也呈现出巨大的优势。尽管将具有自组装功能的萜类小分子作为纳米构建体系应用于协同光化抗癌取代很大的疗效,但传统的制备方法(比如乳化溶剂挥发法)繁琐复杂,且制备的协同抗肿瘤光敏药物缺乏刺激响应释放功能,导致其在生理条件下无法有效的释放活性药物分子发挥疗效,缺乏一定的控制释放能力,另一方面,缺乏刺激响应的复合药物体系可能存在药物突释行为,导致潜在的毒性作用。
技术实现要素:
本发明的目的是提供一种萜类小分子组装的氧化还原响应光敏药物的制备方法,所述光敏药物能够实现肿瘤微环境氧化还原响应释放功能,且安全高效的联合化疗-光动力治疗抗癌。
本发明的目的是通过以下技术方案实现的:
一种萜类小分子组装的氧化还原响应光敏药物的制备方法,包括以下步骤:
步骤一:以天然萜类小分子和二硫代羧酸作为反应物,通过酯缩合反应脱水生成氧化还原功能基二硫修饰的天然萜类小分子;
步骤二:将氧化还原功能基二硫修饰的天然萜类小分子和ce6采用超声共沉淀法制备得到萜类小分子组装的氧化还原响应光敏药物。
相比于现有技术,本发明具有如下优点:
1)采用简单的共组装方式,制备了新型天然萜类小分子介导的无载体纳米复合光敏药物,消除了纳米载体的潜在安全性和抗癌惰性问题;
2)天然萜类小分子的引入,使得纳米复合光敏药物具有优良的生物相容性及生物安全性;
3)天然萜类小分子本身的抗癌活性,进一步实现安全的协同化疗-光动力治疗相结合抗肿瘤治疗的可能;将活性萜类小分子与光动力治疗结合构建协同抗肿瘤纳米复合光敏药物,一方面可以解决纯药光敏剂及萜类小分子本身生物利用度低的缺陷,另一方面基于通透性和滞留效应(epr)提高药物肿瘤靶向性。
4)考虑到肿瘤微环境高浓度谷胱甘肽(gsh)的特征,在天然萜类小分子上枝接具有氧化还原刺激响应的二硫键,能够实现药物的控制释放,降低药物突释引起的毒副作用,提高药物疗效,提高生物利用度。
附图说明
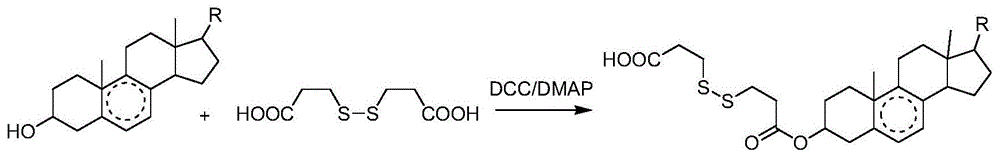
图1为甾体类天然萜类小分子和3,3’-二硫代二丙酸的反应方程式,其中r为≥5个碳原子的直链或者支链烷基,包括饱和烷基和不饱和烷基;
图2为麦角甾醇(ergosterol)分子结构式;
图3为β-谷甾醇(β-sitosterol)分子结构式;
图4为豆甾醇(stigmasterol)分子结构式;
图5为五环三萜类天然萜类小分子和3,3’-二硫代二丙酸反应方程式,其中r为≥0个碳原子的直链或者支链烃基;
图6为五环三萜类天然萜类小分子结构式构型一;
图7为五环三萜类天然萜类小分子结构式构型二;
图8为白桦酯酮酸(betulonicacid)分子结构式;
图9为甘草次酸(glycyrrhetinicacid)分子结构式;
图10为熊果酸(ursolicacid,ua)分子结构式;
图11为白桦脂酸(betulinicacid)分子结构式;
图12为氧化还原功能基二硫修饰的豆甾醇stigmasterol-s-s-cooh的1h-nmr图;
图13为氧化还原功能基二硫修饰的熊果酸ursolicacid-s-s-cooh的1h-nmr图;
图14为uad-ce6nps的扫描电镜(sem)图片;
图15为uad、ce6、uad-ce6nps的紫外-可见吸收光谱;
图16为不同浓度的gsh和ph环境下uad-ce6nps的体外释放曲线,其中,游离的ce6用作对照组,误差线代表标准偏差(n=3);
图17为不同浓度uad-ce6nps在光照与无光照(暗)下4t1细胞存活率图;
图18为荷瘤雌性balb-c老鼠经uad-ce6nps、ce6分别在光照与无光照处理后的肿瘤体积比变化;
图19为荷瘤老鼠经uad-ce6nps药物治疗14天期间的体重变化曲线。
具体实施方式
下面结合附图和具体实施例对本发明的技术方案作进一步的说明,但并不局限于此,凡是对本发明技术方案进行修改或者等同替换,而不脱离本发明技术方案的范围,均应涵盖在本发明的保护范围中。
具体实施方式一
一种萜类小分子组装的氧化还原响应光敏药物的制备方法,包括以下步骤:
步骤一:以天然萜类小分子和二硫代羧酸作为反应物,通过酯缩合反应脱水生成氧化还原功能基二硫修饰的天然萜类小分子;
步骤二:将氧化还原功能基二硫修饰的天然萜类小分子和ce6采用超声共沉淀法制备得到萜类小分子组装的氧化还原响应光敏药物。
进一步的,步骤一中,所述二硫代羧酸的分子结构式为
优选的,步骤一中,所述二硫代羧酸包括2-2’二硫代二乙酸,4-4’二硫代二丁酸,6-6’二硫代二己酸中的一种或多种的组合。
进一步的,步骤一中,向所述反应物中加入二环己基碳二亚胺(dcc)和4-二甲氨基吡啶(dmap)作为催化剂。
进一步的,步骤一中,所述天然萜类小分子和二硫代羧酸的摩尔比为1:1~5。
进一步的,步骤一中,所述天然萜类小分子、dcc和dmap的摩尔比为1:1.2~2.0:1.2~3.0。
进一步的,步骤一中,所述酯缩合反应的时间为15h~48h。
进一步的,步骤一中,向反应物中加入二氯甲烷溶解天然萜类小分子,加入无水n,n-二甲基甲酰胺(dmf)溶解二硫代羧酸,所述天然萜类小分子与二氯甲烷的的比例为0.1-50mg:1ml,所述二硫代羧酸与dmf的比例为0.1-75mg:1ml。
进一步的,步骤一中,酯缩合反应完成后,将反应液进行萃取,然后旋蒸浓缩、真空冷冻干燥,通过正向硅胶柱层析分离纯化得到氧化还原功能基二硫修饰的天然萜类小分子。
进一步的,所述天然萜类小分子包括甾体类天然萜类小分子、五环三萜类天然萜类小分子、三环二萜类天然萜类小分子,但不限于上述三种天然萜类小分子。
进一步的,所述步骤二包括以下步骤:
步骤1、将氧化还原功能基二硫修饰的天然萜类小分子溶解在二甲基亚砜(dmso)中得到样液ⅰ,将ce6溶解在dmso中得到样液ⅱ,所述样液ⅰ的摩尔浓度为10~50mmol/l,所述样液ⅱ的摩尔浓度为10~50mmol/l,取体积比为0.5~8:1的样液ⅰ和样液ⅱ混合均匀得到样液ⅲ;
步骤2、向蒸馏水内加入naoh溶液得到混合溶液ⅰ,在超声作用下,将样液ⅲ加入至混合溶液ⅰ中得到混合溶液ⅱ,其中,混合溶液ⅱ中naoh与ce6的摩尔比为2~4:1,dmso与蒸馏水的体积比为0.2~1:10,将混合溶液ⅱ持续超声0.1~25min后离心,离心后弃去上清液,水洗除去有机试剂得到天然萜类小分子组装的光敏药物。
实施例1:
一种萜类小分子组装的氧化还原响应光敏药物的制备方法,首先构建氧化还原功能基二硫修饰的天然萜类小分子,随后氧化还原功能基二硫修饰的天然萜类小分子与光敏剂ce6通过一个直接、绿色的共组装策略组成而成,制备复合纳米体系即光敏药物。
1)化学合成氧化还原功能基二硫修饰的天然萜类小分子
以天然萜类小分子和3,3’-二硫代二丙酸作为反应物,二环己基碳二亚胺(dcc)和4-二甲氨基吡啶(dmap)做催化剂,通过酯缩合反应脱水生成氧化还原功能基二硫修饰的天然萜类小分子。具体过程如下:
将70mg的天然萜类小分子与3,3’-二硫代二丙酸(摩尔比1:3)加入反应瓶中,再加入6ml二氯甲烷溶解萜类小分子和1ml无水n,n-二甲基甲酰胺(dmf)促进3,3’-二硫代二丙酸的溶解,加入dcc和dmap作为酯缩合反应的脱水剂和催化剂,使摩尔比为萜类小分子:dcc:dmap=1:1.5:2.5,反应进行15h。将反应液用二氯甲烷进行萃取除去未溶解的催化剂与dmf,然后在35-40℃下减压旋蒸浓缩,随后真空冷冻干燥,通过正向硅胶柱层析分离纯化得到氧化还原功能基二硫修饰的天然萜类小分子,石油醚:乙酸乙酯(2:1,v/v)作为洗脱剂。
其中,甾体类天然萜类小分子和3,3’-二硫代二丙酸的反应方程式如图1所示,其中r为≥5个碳原子的直链或者支链烷基,包括饱和烷基和不饱和烷基;优选的甾体类天然萜类小分子为麦角甾醇(ergosterol)、β-谷甾醇(β-sitosterol)、豆甾醇(stigmasterol),其分子结构式如图2-4所示;
五环三萜类天然萜类小分子和3,3’-二硫代二丙酸反应方程式如图5所示,其中r为不少于0个碳原子的直链或者支链烃基;五环三萜类天然萜类小分子存在两种构型,如图6和图7所示;优选的五环三萜类天然萜类小分子为白桦酯酮酸(betulonicacid,bc)、甘草次酸(glycyrrhetinicacid)、熊果酸(ursolicacid)、白桦脂酸(betulinicacid),其分子结构式如图8~图11。
图12为氧化还原功能基二硫修饰的豆甾醇stigmasterol-s-s-cooh的1h-nmr图;通过核磁谱图解析分析,在2.70-2.81ppm出现的两对相同的三重峰(共计4h),以及在2.93ppm处重叠的4个氢的三重峰均为增加的3,3’-二硫代二丙酸上的氢,在4.99-2.37出现的3个氢为豆甾醇母环上双键的三氢,进一步证实甾体类小分子枝接二硫键的合成成功。
图13为氧化还原功能基二硫修饰的熊果酸ursolicacid-s-s-cooh的1h-nmr图;通过核磁氢谱分析,在2.85-2.92ppm出现的两个三重峰共计8h为增加的3,3’-二硫代二丙酸上的亚甲基上的氢,在4.07ppm出现的一个氢为原熊果酸三位羟基碳上的氢,这一结果证实五环三帖类天然小分子枝接二硫键的可以成功合成并制备。
2)以熊果酸(简称ua)为例,制备协同抗肿瘤的光敏药物。
将3,3’-二硫代二丙酸基团修饰的ua简称为uad,将uad与ce6共组装制备的纳米氧化还原响应光敏药物简称uad-ce6nps。
以uad和光敏剂ce6采用超声共沉淀法制备纳米颗粒uad-ce6nps。具体过程如下:
将uad溶解于100μldmso中,配制成22mg/ml(34mm)的样液ⅰ,称取光敏剂二氢卟吩e6(ce6)溶解于100μldmso中,配制成20mg/ml(34mm)的样液ⅱ,分别取20μl样液ⅰ与5μl样液ⅱ混匀(体积比为4:1)。取1ml蒸馏水至青霉素小瓶中,向其中加入10μlnaoh溶液(51mm)得到混合溶液ⅰ,在超声作用下,将uad与ce6的混合液迅速注入青霉素小瓶中得到混合溶液ⅱ,将混合溶液ⅱ持续超声10min后置于台式高速离心机中,转速控制在13000rpm,离心时间为25min。离心后弃上清液,然后水洗一次除去有机试剂得到纳米颗粒uad-ce6nps。
图14为uad与光敏剂ce6制备的纳米材料uad-ce6nps的扫描电镜(sem)图片。采用超声共沉淀法,形成了形貌均一的纳米粒子,且具有优良的分散性,平均粒径尺寸分别为211±4.6nm。
3)uad-ce6nps中光敏剂ce6含量确定
按照步骤2)制备的光敏药物uad-ce6nps物进行后续实验探究。采用紫外-可见分光光度计测定uad-ce6nps中的光敏剂ce6的含量。具体测试方法如下:将步骤2)制备的uad-ce6nps溶于2mldmso使其解组装,ce6游离出来。取制备好的溶液稀释200倍后取2ml装在1cm厚的石英比色皿中并使用紫外-可见分光光度计来分别测量500-800nm波长范围内的紫外-可见光谱。取ce6在665nm特征吸收峰处的吸光值,根据ce6标准曲线计算出uad-ce6nps中的ce6含量。最终确定ce6在uad-ce6nps中的含量为6.2%。
图15为uad、ce6、uad-ce6nps的紫外-可见吸收光谱。组装形成的纳米粒子uad-ce6nps同时拥有典型的uad在260nm附近的吸收峰以及二氢卟吩e6在402nm和655nm处的吸收峰。从ce6相较于分子态的形式而言,在uad-ce6nps水溶液中吸收峰明显展宽、特征峰吸收强度降低,并且最大吸收波长(qy峰669nm)发生红移4nm,表明ce6与uad之间存在明显的π-π相互作用,进一步说明两者通过简单的共组装策略成功制备。
4)uad-ce6nps的体外抗癌活性评估
以鼠源4t1乳腺癌细胞为细胞株,采用标准的mtt法检测uad-ce6nps的化疗活性及bc-ce6nps的光动力抗癌活性。
4t1细胞以4×103个/孔的密度接种96孔板,每孔加100μl细胞悬液,于培养箱中使细胞贴壁生长。每孔加200μl不同浓度梯度的药物共培养24h。实验共分5组,分别为:空白对照组(完全培养基组)、ce6无光照组、ce6光照组、uad组、uad-ce6nps无光照组、uad-ce6nps光照组。其中需要光照的组别在加药4h后,用675±10nm的激光(150mw/cm2)照射各孔10min。所有组别共培养24h后,每孔加100μl0.5mg/mlmtt溶液,4h后每孔加150μldmso,稳定10min后用酶标仪测光密度值,波长为492nm。
5)uad-ce6nps体内抗癌效果评估
以接种4t1肿瘤细胞的balb-c雌性小白鼠为荷瘤研究对象,采用尾静脉注射给药的方式,研究药物抗癌疗效。具体实验分为五组,每组5只老鼠:1.空白对照组:只注射5%葡萄糖水溶液;2.ce6光照组;3.ce6无光照组;4.给药组uad-ce6nps无光照组;5.给药组uad-ce6nps光照组:通过尾静脉注射uad-ce6nps(等当量的ce63.5mg/kg)6h后,对老鼠实施麻醉并将肿瘤红外光下光照处理15min。分别在第0天、3天、6天对老鼠实施相应的治疗14天,期间每隔两天记录一次老鼠肿瘤体积及体重等参数。老鼠肿瘤体积计算公式:v=(l×w2)/2.v代表肿瘤体积,l和w分别代表肿瘤的长和宽。
图16为不同浓度的gsh和ph环境下uad-ce6nps的体外释放曲线(游离的ce6用作对照组,误差线代表标准偏差(n=3)。)在ph7.4和6.5缓冲液中,48h后uad-ce6np中ce6的释放效率分别为58%和63%。在0mmgsh的作用下,药物释放缓慢,48h内只有60%的ce6被释放。在存在1mmgsh和5mmgsh的缓冲液中,与不含gsh的纳米颗粒uad-ce6nps相比,ce6从纳米颗粒uad-ce6nps中的释放明显加速,并且在48h内释放了约75%和80%。由此证明了含二硫键的纳米颗粒uad-ce6nps具有gsh响应性释放。说明二硫键修饰的协同抗肿瘤的光敏药物uad-ce6nps具备氧化还原刺激响应的能力。
图17为不同浓度的uad-ce6nps和单独ce6对4t1细胞的体外毒性结果。图中表明,uad-ce6nps光照组和无光照组对细胞的抑制作用随着浓度的逐渐增加而逐渐增强。相比于uad和ce6无光照组,uad-ce6nps对4t1细胞具有明显的光毒性。在10min的光照下,uad-ce6nps中的2μg/ml等效ce6可能导致4t1细胞死亡约87%,这证实了组装的uad-ce6nps的优异光动力效率。不光照时,uad-ce6nps对4t1细胞具有相对较低的细胞毒性,但在ce6极低毒性下(细胞活力超过71%),它们仍显示出药物浓度依赖性的细胞毒性,这表明uad-ce6nps的这种抗癌活性来自于光敏药物中的具有化疗活性的ua。孵育24h后6.1μg/mluad导致4t1细胞死亡约49%,进一步证实了载体uad的抗癌活性。与单独的游离uad和ce6光动力治疗的活性相比,辐射下的uad-ce6nps具有更有效的联合光动力抗癌效率,对4t1细胞显示出显著的细胞毒性。
图18为荷瘤雌性balb-c老鼠经不同药物光照与无光照处理后的肿瘤体积比变化。无光照的纳米粒子组的小鼠肿瘤体积增长速率小于无光照的ce6组,这表明熊果酸有抑制肿瘤的生物活性;光照ce6组的小鼠肿瘤体积增长速率小于无光照ce6组,这表明pdt疗法在抑制肿瘤中发挥了作用,在相同的ce6给药剂量与同样的光照强度下,纳米颗粒uad-ce6nps组在光照之后肿瘤生长速率缓慢,抑制效果明显优于无光照纳米粒子uad-ce6nps组,证明化学疗法与光动力疗法协同治疗在抑制肿瘤方面的高效性。
图19为进行治疗后各组小鼠的体重变化曲线。除葡萄糖溶液组的小鼠体重随着时间以及肿瘤体积逐步增加,体重亦随之增加外,其它的治疗组增长曲线规律相似,均没有导致小鼠体重下降。结果表明自组装形成的纳米粒子uad-ce6nps在对小鼠的治疗过程中对小鼠的生长发育没有显著的影响,证明制备的纳米粒子uad-ce6nps是低毒安全的。
- 还没有人留言评论。精彩留言会获得点赞!