本发明涉及一种吡喃衍生物药学上可接受的盐或其盐的水合物,以及其晶型结构、制备方法或其药物组合物和它们在制备二肽激酶-IV(DPP-IV)抑制剂药物上的用途。
背景技术:
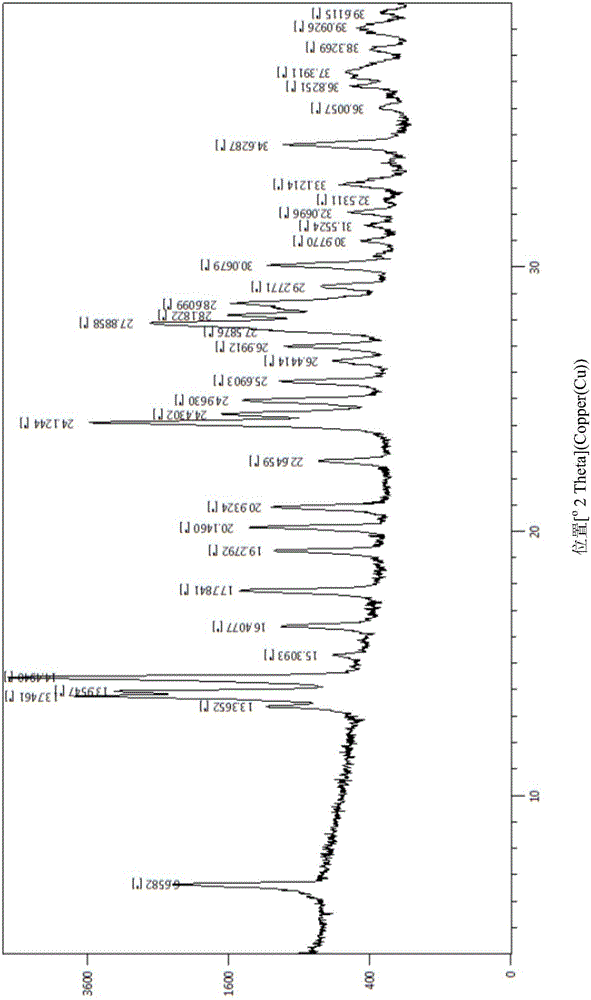
:二肽基肽酶-IV(DipeptidylPeptidase,DPP-IV,EC3.4.14.5)是一个丝氨酸蛋白酶,从含有L-脯氨酸和L-丙氨酸的多肽N端倒数第二位水解N端二肽。通过增强肠促胰岛素活性发挥作用,属于非胰岛素治疗药物。DPP-IV抑制剂无体重增加和水肿等不良反应。PCT/CN2015/080570专利申请公开了新的吡喃衍生物(2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯并[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃基-3-胺,结构式如式(A)。该结构显示对DPP-IV具有良好的抑制作用,具有用于预防和/或治疗II型糖尿病的潜能。技术实现要素:本发明主要目的在于提供吡喃衍生物:(2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯并[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃基-3-胺的盐或其盐的水合物及其晶型。在本发明药物或其组合物的制备中,其盐或其盐的水合物及其晶型比游离碱化合物具有更好的稳定性,耐高温、高湿及强光照,使其更适于药物剂型的制备。本发明涉及一种式(I)所示化合物的盐或其盐的水合物:本发明优选方案,式(I)所示化合物的盐或其盐的水合物为式(II)结构:其中,HA为药学上可接受的酸,比如盐酸、硫酸、磷酸、醋酸、丙酸、丙二酸、草酸、己二酸、三氟乙酸、琥珀酸、水杨酸、苯甲酸、邻苯二甲酸、乳酸、酒石酸、苹果酸、苯磺酸、甲磺酸、对甲苯磺酸、柠檬酸或富马酸;n选自1或2;m选自0~3。本发明优选方案,HA选自盐酸、硫酸、磷酸、醋酸、丙酸、丙二酸、草酸、己二酸、三氟乙酸、琥珀酸、水杨酸、苯甲酸、邻苯二甲酸、乳酸、酒石酸、苹果酸、苯磺酸、甲磺酸、对甲苯磺酸、柠檬酸或富马酸。本发明优选方案,当HA选自盐酸、磷酸或对甲苯磺酸时,n选自1或2;当HA选自苯甲酸、柠檬酸或富马酸时,n选自1。本发明优选方案,一种式(III)所示的化合物:其中,m选自1~3。本发明优选方案,式(III)所示的化合物为晶型I,使用Cu-Kα辐射,其X-射线粉末衍射图谱在以下2θ位置具有特征衍射峰:6.7°±0.2°、13.8°±0.2°、14.5°±0.2°、24.2°±0.2°和27.9°±0.2°。本发明优选方案,式(III)所示的化合物为晶型I,使用Cu-Kα辐射,其X-射线粉末衍射图谱还在以下2θ位置具有特征衍射峰:14.0°±0.2°、16.5°±0.2°、17.8°±0.2°、19.3°±0.2°、20.3°±0.2°和21.0°±0.2°。本发明优选方案,式(III)所示的化合物为晶型I,使用Cu-Kα辐射,其X-射线粉末衍射图谱还在以下2θ位置具有特征衍射峰:13.4°±0.2°、15.4°±0.2°、22.7°±0.2°、24.5°±0.2°和25.0°±0.2°。本发明优选方案,式(III)所示的化合物的晶型I的X-射线粉末衍射图谱基本如图1所示。本发明优选方案,式(III)所示的化合物的差示扫描量热热分析图基本如图2所示。本发明优选方案,式(III)所示的化合物的热重分析曲线基本如图3所示。本发明优选方案,式(III)所示的化合物,m=3。本发明还提供一种药物组合物,其包含治疗有效量的如上所述盐或其盐的水合物,以及药学上可接受的载体或赋形剂。本发明还提供通式(I)所示的化合物的盐或其盐的水合物,或通式(II)或通式(III)所述化合物,或如上所述的药物组合物在制备用于预防和/或治疗糖尿病和/或糖尿病并发症的药物中的应用。优选地,所述糖尿病并发症包括糖尿病性视网膜病、糖尿病性神经病和糖尿病性肾病中的一种或多种。优选地,所述糖尿病包括II型糖尿病。本发明还提供一种治疗代谢性疾病的方法,所述方法包括给药本发明任意所述的盐或其盐的水合物,或前述药物组合物。本发明化合物的合成方法为了完成本发明的目的,本发明化合物可以由以下方案制备而得:式(IV)化合物与HA反应得到式(II);或式(V)化合物与HA反应得到式(II);其中,HA选自盐酸、硫酸、磷酸、醋酸、丙酸、丙二酸、草酸、己二酸、三氟乙酸、琥珀酸、水杨酸、苯甲酸、邻苯二甲酸、乳酸、酒石酸、苹果酸、苯磺酸、甲磺酸、对甲苯磺酸、柠檬酸或富马酸;n选自1或2;m选自0~3;P为氨基保护基。优选地,HA选自盐酸、磷酸、苯甲酸、对甲苯磺酸、柠檬酸或富马酸。更优选地,HA为质量分数为15%~37%的盐酸。通式(IV)化合物或者通式(V)化合物可用溶剂溶解后,加入HA酸进行反应,其中:溶剂可以是一切能够溶解通式(IV)化合物或者通式(V)化合物的溶剂,比如乙醇;反应在0~90℃均可以。反应得到的产物纯度高,作为选择,得到的产物还可以进一步重结晶。重结晶时,产物可以选择离子水与乙醇的混合液溶解,加热溶解后,用合适的析晶溶剂析出产物,所述的析晶溶剂,比如醋酸异丙酯、乙酸乙酯、异丙醚或甲基叔丁基醚。本发明的反应根据TLC或者HPLC等本领域技术人员常用的方法跟踪反应进度。本发明二盐酸盐水合物合成中,盐酸与游离碱化合物的摩尔比例为3:1至10:1。本发明中参与反应的酸一般在0℃~室温条件下滴加到反应体系,可以在0℃以下滴加。除非有相反的陈述,本发明中使用的术语具有下述含义。本发明所述基团和化合物中所涉及的碳、氢、氧、硫、氮或卤素均包括它们的同位素,及本发明所述基团和化合物中所涉及的碳、氢、氧、硫、氮或卤素任选进一步被一个或多个它们对应的同位素所替代,其中碳的同位素包括12C、13C和C14,氢的同位素包括氕(H)、氘(D,又称为重氢)、氚(T,又称为超重氢),氧的同位素包括16O、17O和18O,硫的同位素包括32S、33S、34S和36S,氮的同位素包括14N和15N,氟的同位素19F,氯的同位素包括35Cl和37Cl,溴的同位素包括79Br和81Br。本文所述化合物可以含有一个或多个不对称中心,并且由此可以以外消旋物、外消旋混合物、单一对映异构体、非对映异构体混合物和单一非対映异构体存在。“有效剂量“指引起组织、系统或受试者生理或医学翻译的化合物的量,此量是所寻求的,包括在受治疗者身上施用时足以预防受治疗的疾患或病症的一种或几种症状发生或使其减轻至某种程度的化合物的量。“IC50”指半数抑制浓度,指达到最大抑制效果一半时的浓度。数值范围a~b表示从数a到数b的任意数,比如,“m选自1~3”表示m可以选自1至3任意的数值。本发明晶型结构可以使用本领域普通技术人员已知的各种分析技术分析,包括但不限于,X-射线粉末衍射(XRD)、示差扫描热法(DSC)和/或热重法(TG)。热重分析(ThermogravimetricAnalysis,TGA),又叫热重法(Thermogravimetry,TG)。附图说明图1是化合物1在80℃干燥16小时所得的晶型I使用Cu-Kα辐射的X-射线粉末衍射图谱。图2是化合物1在80℃干燥16小时所得的晶型I的差示扫描量热(DSC)热分析图。图3是化合物1在80℃干燥16小时所得的晶型I的热重(TG)曲线-微熵热重分析(DTG)曲线。图4是化合物1在80℃干燥8小时所得的晶型I使用Cu-Kα辐射的X-射线粉末衍射图谱。图5是化合物1在60℃干燥10小时所得的晶型I使用Cu-Kα辐射的X-射线粉末衍射图谱。图6是化合物1在30℃干燥8小时所得的晶型I使用Cu-Kα辐射的X-射线粉末衍射图谱。图7A是由单晶衍射确定的化合物1的单晶中分子球棍模型;图7B为与图7A相对应的由单晶确定的绝对构型;图7C为单晶中氢键示意图(图中O2~O4表示水分子)。图8是实施例9中化合物1重结晶前的晶型I使用Cu-Kα辐射的X-射线粉末衍射图谱。图9是实施例9中化合物1重结晶后的晶型使用Cu-Kα辐射的X-射线粉末衍射图谱。具体实施方式以下通过具体实施例详细说明本发明的实施过程和产生的有益效果,旨在帮助阅读者更好地理解本发明的实质和特点,不作为对本案可实施范围的限定。化合物的结构是通过核磁共振(NMR)和/或质谱(MS)来确定的。NMR的测定是用(BrukerADVANCEIII400)核磁仪,测定溶剂为氘代二甲基亚砜(DMSO-d6),氘代氯仿(CDCl3),氘代甲醇(CD3OD),内标为四甲基硅烷(TMS)。MS的测定用(Agilent6120B(ESI))。HPLC的测定使用安捷伦1260DAD高压液相色谱仪(Zorba×SB-C18100×4.6mm)。薄层层析硅胶板使用烟台黄海HSGF254或青岛GF254硅胶板,薄层色谱法(TLC)使用的硅胶板采用的规格是0.15mm~0.20mm,薄层层析分离纯化产品采用的规格是0.4mm~0.5mm。柱层析一般使用烟台黄海硅胶200~300目硅胶为载体。无特殊说明,甲基叔丁基醚、四丁基溴化铵、氢化钠、三氟乙酸购买于成都市科龙化工试剂厂;二碳酸二叔丁基酯、N,N'-二羰基二咪唑、N,O-二甲基羟氨盐酸盐购买于爱斯特(成都)医药技术有限公司;碳酸铯、N-羟基丁二酰亚胺、二苯亚甲基甘氨酸乙酯购买于安耐吉化学;2,5-二氟溴苯购买于上海德默医药科技有限公司;异丙基氯化镁/氯化锂四氢呋喃溶液购买于百灵威科技有限公司;炔丙基苯磺酸酯、四丁基氟化铵、三(乙酰氧基)硼氢化钠、四丁基六氟磷酸胺购买于阿达玛斯试剂公司;环戊二烯基双(三苯基膦)氯化钌(II)购买于ACROSorgainics;硼烷二甲硫醚购买于韶远化学科技(上海)有限公司;过硼酸钠购买于天津光复精细化工研究所;氯{[(1R,2R)-(-)-2-氨基-1,2-二苯基乙基](五氟苯磺酰)氨基}(对伞花烃)钌(II)购买于Stremchemical;碘甲烷购买于国药集团药业股份有限公司。氮气氛是指反应瓶连接一个约1L容积的氮气气球。氢气氛是指反应瓶连接一个约2L容积的氢气气球。氢化反应通常抽真空,充入氢气,反复操作3次。实施例中无特殊说明,溶液是指水溶液。实施例中无特殊说明,反应的温度为室温。室温温度为10℃~30℃。中间体1:((2R,3S)-2-(2,5-二氟苯基)-5-羰基四氢-2H-吡喃-3-基)氨基甲酸叔丁酯(中间体1)tert-butyl((2R,3S)-2-(2,5-difluorophenyl)-5-oxotetrahydro-2H-pyran-3-yl)carbamate第一步:2-氨基戊基-4-炔酸乙酯(1B)ethyl2-aminopent-4-ynoate室温下,将1A(50g,0.187mol)溶于甲基叔丁基醚(300mL)中,加入炔丙基苯磺酸酯(44g,0.224mol)和四丁基溴化铵(6.1g,0.019mol),升温至50℃,加入碳酸铯(121.8g,0.374mol),于50℃温度下反应过夜。将反应液过滤,滤饼用甲基叔丁基醚(40mL×2)洗涤,合并有机相,旋蒸浓缩至一半体积,加入盐酸溶液(3mol/L,100mL),室温下搅拌1小时,静置分液,水相用甲基叔丁基醚(70mL×2)萃取,收集水相,浓缩得到1B。第二步:2-((叔丁氧羰基)氨基)-4-炔戊酸(1C)2-((tert-butoxycarbonyl)amino)pent-4-ynoicacid将氢氧化钠(33.7g,0.842mol)溶于水(100mL),逐滴滴加至1B(26.4g,0.187mol)中,室温下搅拌2小时,滴加二碳酸二叔丁基酯(45g,0.206mol)的甲基叔丁基醚(125mL)溶液,室温下搅拌4小时。静置分液,水相用甲基叔丁基醚(80mL×2)洗涤,水相用3mol/L的盐酸溶液调节pH约3,用甲基叔丁基醚(100mL×2)萃取,合并有机相,有机相用饱和氯化钠水溶液(30mL×2)洗涤,无水硫酸镁干燥,过滤,旋干,得到黄色油状液体1C(33g,产率83%)。Msm/z(ESI):212.0[M-1]。第三步:(1-(甲氧基(甲基)氨基)-1-羰基戊基-4-炔-2-基)氨基甲酸叔丁酯(1D)tert-butyl(1-(methoxy(methyl)amino)-1-oxopent-4-yn-2-yl)carbamate将1C(33g,0.155mol)溶于N,N-二甲基甲酰胺(200mL)中,控制温度小于10℃,加入N,N'-羰基二咪唑(32.58g,0.201mol),0℃下反应1小时,加入N,O-二甲基羟氨盐酸盐(19.6g,0.186mol),室温搅拌过夜。向反应液中逐滴加入水(150mL),搅拌1小时,用乙酸乙酯(100mL×2)萃取,合并有机相,有机相依次用饱和碳酸氢钠溶液(60mL×3)、饱和氯化钠溶液(60mL×3)洗涤,无水硫酸镁干燥,过滤,浓缩,残留物用硅胶柱色谱分离提纯(石油醚/乙酸乙酯(v/v)=10:1),得到白色固体1D(35g,产率88.2%)。Msm/z(ESI):156.9[M-100]。第四步:(1-(2,5-二氟苯基)-1-羰基戊基-4-炔-2-基)氨基甲酸叔丁酯(1E)tert-butyl(1-(2,5-difluorophenyl)-1-oxopent-4-yn-2-yl)carbamate氮气保护下,将2,5-二氟溴苯(15.05g,78mmol)溶于干燥甲苯(50mL)中,冰盐浴降温至-10℃以下,逐滴加入异丙基氯化镁/氯化锂四氢呋喃溶液(66mL,1.3mol/L),保持在-10℃左右搅拌1小时,滴加1D(10g,39mmol)的干燥四氢呋喃(100mL)溶液,保持温度-10℃,加毕,于室温下反应4小时,将温度降至-10℃左右,逐滴加入饱和氯化铵溶液(40mL),搅拌10分钟,用3mol/L的盐酸溶液调节pH5~6,静置分液,水相用甲基叔丁基醚(50mL×2)萃取,合并有机相,有机相用饱和氯化钠溶液(30mL×2)洗涤,无水硫酸钠干燥,过滤,浓缩,残留物用硅胶柱色谱分离提纯(石油醚/乙酸乙酯(v/v)=50:1~8:1),得到淡黄色固体1E(10.1g,产率83.5%)。Msm/z(ESI):210.1[M-100]。第五步:((1R,2S)-1-(2,5-二氟苯基)-1-羟基戊基-4-炔-2-基)氨基甲酸叔丁酯(1F)tert-butyl((1R,2S)-1-(2,5-difluorophenyl)-1-hydroxypent-4-yn-2-yl)carbamate将1E(16.07g,52mmol)溶于四氢呋喃(100mL),加入三乙烯二胺(17.39g,155mmol)与氯{[(1R,2R)-(-)-2-氨基-1,2-二苯基乙基](五氟苯磺酰)氨基}(对伞花烃)钌(II)(即RuCl(p-cymene)(R,R)-FSDPEN)(0.37g,0.52mmol),逐滴加入甲酸(14.27g,310mmol),加毕,于40℃反应过夜。将反应液浓缩除去四氢呋喃和甲酸,向残留物中加入水(60mL)和盐酸(3mol/L,10mL),用甲基叔丁基醚(90mL×3)萃取,合并有机相,有机相用饱和碳酸氢钠溶液(35mL×2)洗涤,无水硫酸镁干燥,过滤,浓缩,残留物用硅胶柱色谱分离提纯(石油醚/乙酸乙酯(v/v)=60:1~10:1),得到淡黄色胶状物1F(15.37g,产率95%)。Msm/z(ESI):334.2[M+23]。第六步:((2R,3S)-2-(2,5-二氟苯基)-3,4-二氢-2H-吡喃-3-基)氨基甲酸叔丁酯(1G)tert-butyl((2R,3S)-2-(2,5-difluorophenyl)-3,4-dihydro-2H-pyran-3-yl)carbamate将1F(15.37g,49.4mmol)加热条件下溶于N,N-二甲基甲酰胺(75mL),加入四丁基六氟磷酸胺(2.49g,6.42mmol)、N-羟基丁二酰亚胺(2.84g,24.75mmol)、三苯基膦(0.86g,3.26mmol)和碳酸氢钠(2.16g,25.69mmol),氮气置换三次,抽真空15分钟,加入环戊二烯基双(三苯基膦)氯化钌(II)(即CpRuCl(PPh3)2)(1.79g,2.47mmol),氮气置换三次,并抽真空15分钟,氮气保护下,升温至85℃反应过夜。向反应液中加入水(300mL)和甲基叔丁基醚(200mL),用硅胶过滤,滤液静置后分液,水相用甲基叔丁基醚(90mL×2)萃取,合并有机相,有机相用饱和碳酸氢钠溶液(60mL×2)洗涤,无水硫酸钠干燥,过滤浓缩,残留物用硅胶柱色谱分离提纯(石油醚/乙酸乙酯(v/v)=80:1~30:1),得到淡黄色粉末固体1G(8.9g,产率57.9%)。Msm/z(ESI):256.2[M-56]。第七步:((2R,3S)-2-(2,5-二氟苯基)-5-羟基四氢-2H-吡喃-3-基)氨基甲酸叔丁酯(1H)tert-butyl((2R,3S)-2-(2,5-difluorophenyl)-5-hydroxytetrahydro-2H-pyran-3-yl)carbamate将1G(8.9g,28.6mmol)溶解于干燥甲基叔丁基醚(90mL)中,加入干燥甲苯(9mL),温度降至-10℃,逐滴加入硼烷二甲硫醚四氢呋喃溶液(2mol/L,35.9mL),于0℃下反应3.5小时。缓慢加入水(4mL),逐滴加入氢氧化钠溶液(1mol/L,89mL),搅拌15分钟,分批加入过硼酸钠(13.2g,85.8mmol),室温搅拌过夜。静置分液,水相用甲基叔丁基醚(50mL×2)萃取,合并有机相,有机相用饱和氯化钠溶液(20mL×2)洗涤,无水硫酸钠干燥,过滤,浓缩,向残留物中加入甲苯(50mL),加热至90℃溶解,将正己烷(200mL)滴加至反应液中,析出白色固体,过滤,正己烷(30mL×2)洗涤滤饼,浓缩除去溶剂,得到白色固体粉末1H(7.9g,产率84%)。Msm/z(ESI):274.1[M-56]。第八步:((2R,3S)-2-(2,5-二氟苯基)-5-羰基-四氢-2H-吡喃-3-基)氨基甲酸叔丁酯(中间体1)tert-butyl((2R,3S)-2-(2,5-difluorophenyl)-5-oxo-tetrahydro-2H-pyran-3-yl)carbamate将1H(11.53g,35.03mmol)溶解于二氯甲烷(130mL),降温至0℃,将戴斯马丁氧化剂(29.72g,70.06mmol)分批加至反应液中,自然升至室温反应4小时。降温至0℃,将饱和碳酸氢钠溶液(60mL)滴加至反应液中,搅拌20分钟,过滤,滤液静置分液,水相用甲基叔丁基醚(60mL×3)萃取,合并有机相,有机相用饱和碳酸氢钠溶液(30mL×2)洗涤,无水硫酸钠干燥,过滤浓缩,残留物用硅胶柱色谱分离提纯(石油醚/乙酸乙酯(v/v)=10:1-4:1),得到白色晶状粉末中间体1(10.85g,产率94.7%)。Msm/z(ESI):272.0[M-56];1HNMR(400MHz,DMSO-d6):δ7.29-7.13(m,4H),4.77–4.75(d,1H),4.22-4.12(d,2H),4.08-4.02(m,1H),2.75-2.70(m,2H),1.23(s,9H)。实施例1(2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯并[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃-3-胺二盐酸盐水合物(化合物1)(2R,3S,5R)-2-(2,5-difluorophenyl)-5-(1-methyl-2-(1-methyl-1H-tetrazol-5-yl)pyrrolo[3,4-d]imidazol-5(1H,4H,6H)-yl)tetrahydro-2H-pyran-3-aminedihydrochloridehydrate第一步:2-乙氧基-2-亚氨基乙酸乙酯盐酸盐(1b)ethyl2-ethoxy-2-iminoacetatehydrochloride将氰基甲酸乙酯1a(9.9g,0.1moL)溶于干燥的乙醚(50mL)中,并加入干燥的无水乙醇(5.52g,0.12moL),冰浴搅拌下通入干燥的氯化氢气体直至饱和(体系增重5g)并继续搅拌2小时,过滤析出的白色固体,滤饼用无水乙醚(20mL×3)洗涤,真空干燥,得到白色固体1b(14g,产率77%)。第二步:5-叔丁基2-乙基3a,4,6,6a-四氢吡咯并[3,4-d]咪唑-2,5(1H)-二甲酸酯(1c)5-tert-butyl2-ethyl3a,4,6,6a-tetrahydropyrrolo[3,4-d]imidazole-2,5(1H)-dicarboxy-late将叔丁基3,4-二胺基吡咯烷-1-羧酸酯盐酸盐(980mg,4.126mmoL)溶于六氟异丙醇(10mL)中,搅拌下加入1b(886mg,4.538mmoL),50℃搅拌16小时。将反应液浓缩旋干,加入饱和食盐水(50mL),用稀盐酸调节pH值至2~3,用乙酯乙酯(50mL×2)萃取。收集水相,并用饱和碳酸氢钠溶液调pH值7~8,用乙酯乙酯(50mL×4)萃取,合并有机相,无水硫酸钠干燥,过滤,浓缩,柱层析分离纯化(二氯甲烷/甲醇(v/v)=20:1),得到白色固体1c(700mg,产率60%)。MSm/z(ESI):284.1[M+1];1HNMR(400MHz,CDCl3):δ4.64(S,2H),4.37-4.33(q,2H),3.72-3.69(d,2H),3.55-3.50(dd,2H),1.44(s,9H),1.40-1.37(t,3H)。第三步:5-叔丁基2-乙基4,6-二氢吡咯并[3,4-d]咪唑-2,5(1H)-二甲酸酯(1d)(2R,4R)-tert-butyl4-(tert-butyldimethylsilyloxy)-2-(methoxymethyl)pyrrolidine-1-carboxylate将草酰氯(652.5mg,5.14mmoL)溶于干燥的二氯甲烷(15mL)中,用干冰丙酮浴降温到-78℃,搅拌下滴加干燥的二甲基亚砜(803.96mg,10.29mmoL),搅拌30分钟。向反应液中滴加1c(970mg,3.43mmoL)的二氯甲烷(5mL)溶液,继续搅拌20分钟。-78℃下滴加二异丙基乙胺(2.216g,17.15mmoL),自然升到室温反应2小时。向反应液中加入饱和氯化铵溶液(50mL)、饱和氯化钠溶液(50mL)和二氯甲烷(50mL),分液,水相用二氯甲烷用(50mL×3)萃取,合并有机相,饱和氯化钠溶液(50mL×3)洗涤,无水硫酸钠干燥,过滤,浓缩,柱层析分离纯化(石油醚/乙酸乙酯=1:1)得到白色固体1d(520mg,产率54%)。MSm/z(ESI):282.1[M+1];1HNMR(400MHz,CDCl3):δ4.54(s,2H),4.47(s,2H),4.46-4.41(q,2H),1.51(s,9H),1.44-1.41(t,3H)。第四步:5-叔丁基2-乙基1-甲基-4,6-二氢吡咯并[3,4-d]咪唑-2,5(1H)-二甲酸酯(1e)5-tert-butyl2-ethyl1-methyl-4,6-dihydropyrrolo[3,4-d]imidazole-2,5(1H)-dicarboxylate将1d(5.4g,19.2mmoL)溶于N,N-二甲基甲酰胺(110mL)中,加入碳酸钾(3.3g,23.9mmoL),冰浴下加入碘甲烷(3.4g,23.9mmoL),室温反应2小时,向反应液中加入饱和氯化铵溶液(300mL),及饱和氯化钠溶液(200mL),用乙酸乙酯(200mL×3)萃取,合并有机相,用饱和氯化钠溶液(200mL×3)洗涤。用无水硫酸钠干燥,过滤,将滤液减压浓缩,得到浅黄色固体1e(5.3g,产率94%)。第五步:叔丁基2-氨甲酰基-1-甲基-4,6-二氢吡咯并[3,4-d]咪唑-5(1H)-甲酸酯(1f)tert-butyl2-carbamoyl-1-methyl-4,6-dihydropyrrolo[3,4-d]imidazole-5(1H)-carboxy-late将1e(2.3g,7.8mmol),溶于氨-甲醇溶液(40mL,7mol/L,280mmol)中,升温至85℃封管搅拌反应20小时。将反应液冷却至室温,过滤,滤饼用乙酸乙酯(10mL×3)洗涤,滤液浓缩后与滤饼合并,得到白色固体1f(1.914g,产率93%)。第六步:叔丁基2-氰基-1-甲基-4,6-二氢吡咯并[3,4-d]咪唑-5(1H)-甲酸酯(1g)tert-butyl2-cyano-1-methyl-4,6-dihydropyrrolo[3,4-d]imidazole-5(1H)-carboxylate将1f(1.576g,5.925mmol),溶于N,N-二甲基甲酰胺(45mL)中,加入五氯化磷(1.482g,7.109mmol),升温至50℃反应2小时。将反应液冷却至室温,加入乙酸乙酯(50mL),饱和氯化钠溶液(150mL),分液,水相用乙酸乙酯(50mL×3)萃取,合并有机相,分别用饱和食盐水(50mL×3)、饱和碳酸氢钠溶液(50mL×2)洗涤,无水硫酸钠干燥,过滤,将滤液浓缩得到白色固体1g(1.25g,产率85%)。第七步:叔丁基1-甲基-2-(1H-四唑-5-基)-4,6-二氢吡咯并[3,4-d]咪唑-5(1H)-甲酸酯(1h)tert-butyl1-methyl-2-(1H-tetrazol-5-yl)-4,6-dihydropyrrolo[3,4-d]imidazole-5(1H)-carboxylate将1g(1030mg,4.153mmoL)溶于N,N-二甲基甲酰胺(20mL)中,加入叠氮化钠(810mg,12.459mmoL)及醋酸铵(960mg,12.459mmoL),升温至120℃反应过夜。加入1mol/L盐酸溶液调节pH值1~2,加水(200mL),过滤,滤液用二氯甲烷(50mL×3)萃取。合并有机相,用无水硫酸钠干燥,过滤,旋干,烘干得到黄色固体1h(1000mg,产率83%)。MSm/z(ESI):292.1[M+1]。第八步:叔丁基1-甲基-2-(2-甲基-2H-四唑-5-基)-4,6-二氢吡咯并[3,4-d]咪唑-5(1H)-甲酸酯(1i-1)tert-butyl1-methyl-2-(2-methyl-2H-tetrazol-5-yl)-4,6-dihydropyrrolo[3,4-d]imidazole-5(1H)-carboxylate叔丁基1-甲基-2-(1-甲基-1H-四唑-5-基)-4,6-二氢吡咯并[3,4-d]咪唑-5(1H)-甲酸酯(1i-2)tert-butyl1-methyl-2-(1-methyl-1H-tetrazol-5-yl)-4,6-dihydropyrrolo[3,4-d]imidazole-5(1H)-carboxylate将1h(1000mg,3.436mmoL)溶于N,N-二甲基甲酰胺(25mL)中,冰浴下加入氢化钠(165mg,4.124mmoL),冰浴下搅拌0.5小时后加入碘甲烷(586mg,4.124mmoL),自然升温反应过夜。将反应液缓慢加入碎冰(50g)中,加入氯化钠使溶液饱和,乙酸乙酯(50mL×4)萃取。合并有机相,用饱和氯化钠溶液(50mL×3)洗涤,用无水硫酸钠干燥,过滤,旋干,残留物用硅胶柱层析分离纯化(石油醚/乙酸乙酯(v/v)=2:1~1:1),得到白色固体1i-1(682mg,产率55%)和白色固体1i-2(230mg,产率18%)。MSm/z(ESI):306.1[M+1]。1i-1:1HNMR(400MHz,DMSO):δ4.58-4.54(d,2H),4.39-4.37(d,2H),4.33(s,3H),3.99(s,3H),1.47(s,9H)。1i-2:1HNMR(400MHz,DMSO):δ4.54-4.50(d,2H),4.44(s,3H),4.34-4.31(d,2H),3.92-3.91(m,3H),1.47(s,9H)。第九步:1-甲基-2-(1-甲基-1H-四唑基-5-基)-1,4,5,6-四氢吡咯并[3,4-d]咪唑二苯磺酸盐(1j)1-methyl-2-(1-methyl-1H-tetrazol-5-yl)-1,4,5,6-tetrahydropyrrolo[3,4-d]imidazoledibenzenesulfonate将1i-1(682mg,2.236mmoL)、苯磺酸.1.5H2O(827.3mg,4.472mmoL)溶于二氯甲烷(25mL)中,升温至40℃反应过夜。将反应液浓缩旋干,真空干燥得到1j(1165mg),直接用下一步反应。MSm/z(ESI):206.1[M+1]。第十步:叔丁基((2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯并[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃-3-基)氨基甲酸酯(1k)tert-butyl((2R,3S,5R)-2-(2,5-difluorophenyl)-5-(1-methyl-2-(1-methyl-1H-tetrazol-5-yl)pyrrolo[3,4-d]imidazol-5(1H,4H,6H)-yl)tetrahydro-2H-pyran-3-yl)carbamate将1j(1165mg,2.236mmoL)及中间体1(804.4mg,2.459mmoL)溶于N,N-二甲基乙酰胺(10mL)中,室温下搅拌0.5小时,冰浴下加入三(乙酰氧基)硼氢化钠(1279mg,6.037mmoL),0℃搅拌0.5小时后自然升至室温搅拌2小时。补加中间体1(400mg,1.223mmoL)及三(乙酰氧基)硼氢化钠(400mg,1.887mmoL),室温反应2小时。将反应液加入饱和碳酸氢钠溶液(100mL)中,搅拌0.5小时,过滤,滤饼用水(20mL×3)洗涤后用二氯甲烷(100mL)溶解,用无水硫酸钠干燥,过滤,旋干,残留物用硅胶柱层析分离纯化(二氯甲烷/甲醇(v/v)=60:1),得到的粗产品用乙醚(50mL)溶解,加入正己烷(100mL),过滤,得到黄色固体1k(480mg,产率42%)。MSm/z(ESI):517.1[M+1]。第十一步:(2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯并[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃-3-胺(1l)(2R,3S,5R)-2-(2,5-difluorophenyl)-5-(1-methyl-2-(1-methyl-1H-tetrazol-5-yl)pyrrolo[3,4-d]imidazol-5(1H,4H,6H)-yl)tetrahydro-2H-pyran-3-amine将1k(440mg,0.853mmoL)加入二氯甲烷(5mL)中,冰浴下加入三氟乙酸(2mL),自然升至室温反应2小时。将反应液旋干,加入饱和碳酸氢钠溶液(100mL)调节pH值7~8,水相用二氯甲烷(30mL×4)萃取。合并有机相,用无水硫酸钠干燥,过滤,旋干,残留物用硅胶柱层析分离纯化(二氯甲烷/甲醇(v/v)=30:1,加入少量氨水),得到黄色固体1l(134mg,产率38%)。MSm/z(ESI):417.1[M+1];1HNMR(400MHz,CD3OD):δ7.23-7.06(m,3H),4.38(s,3H),4.32-4.26(m,2H),4.07-4.05(t,2H),4.03(s,3H),3.95-3.94(t,2H),3.46-3.41(t,1H),3.17-3.09(m,1H),2.96-2.90(m,1H),2.51-2.47(m,1H),1.58-1.49(q,1H)。第十二步:(2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯并[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃-3-胺二盐酸盐水合物(化合物1)(2R,3S,5R)-2-(2,5-difluorophenyl)-5-(1-methyl-2-(1-methyl-1H-tetrazol-5-yl)pyrrolo[3,4-d]imidazol-5(1H,4H,6H)-yl)tetrahydro-2H-pyran-3-aminedihydrochloridehydrate1l(16g,38.43mmol)和无水乙醇(80mL)依次加入反应瓶中,将浓盐酸(8mL)滴加到反应液中,加毕于室温搅拌10分钟,反应液中固体溶解完全,搅拌10分钟后析出浅白色固体。将醋酸异丙酯(56mL)滴加到反应液中,室温继续搅拌1小时。将反应液过滤,滤饼用醋酸异丙酯(30mL×2)洗涤,抽干,真空干燥,得到灰白色固体化合物1(14g,收率85%)。图2是所得化合物1在80℃干燥16小时所得的晶型I的差示扫描量热(DSC)热分析图。图3是化合物1在80℃干燥16小时所得的晶型I的热重(TG)曲线-微熵热重分析(DTG)曲线。化合物1通过单晶衍射测定化合物1所含HCl个数为2。单晶培养:取约5mg样品放于玻璃小瓶中,用300μL乙醇超声溶清,室温下小孔挥发,得到小块状晶体,将此晶体作为晶种加至样品的乙醇饱和溶液中,小孔挥发,得到块状晶体。单晶衍射仪器信息和检测方法参数如表1所示:表1热台偏正光显微镜信息和检测方法参数如表2所示:表2所得晶胞参数如下表3所示:表3单晶结构信息表图7A是由单晶衍射确定的化合物1的单晶中分子球棍模型;图7B为与图7A相对应的由单晶确定的绝对构型;图7C为单晶中氢键示意图(图中O2~O4表示水分子),从图7A~7C可知,一个API分子结合3个水分子,其中含O2的水分子与API分子通过氢键连接,比较稳定,为结晶水;含O3和O4的两个水分子处于游离的状态,容易失去,但是对晶胞性状不影响。实施例2(2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯并[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃-3-胺二盐酸盐水合物(化合物1)(2R,3S,5R)-2-(2,5-difluorophenyl)-5-(1-methyl-2-(1-methyl-1H-tetrazol-5-yl)pyrrolo[3,4-d]imidazol-5(1H,4H,6H)-yl)tetrahydro-2H-pyran-3-aminedihydrochloridehydrate室温下,1k(133g,0.257mol)和乙醇(864.5mL)依次加入反应瓶中,将浓盐酸(146.3mL)滴加到反应液中,加毕于60℃~70℃反应3小时。将醋酸异丙酯(296.2mL)滴加到反应液中,自然冷却到室温,析出浅白色固体。过滤,滤饼用醋酸异丙酯(300mL×2)洗涤,抽干,真空干燥,得到灰白色固体化合物1(102g,收率80%)。实施例1或实施例2中化合物1因真空干燥的温度、时间不同,得到的水合物所含的水分子含量也不同,即包含的水分子个数不同,m值不同,见表4。表4化合物1经不同温度和干燥时间干燥后的含水量图1所示为80℃干燥温度、干燥16小时的X-粉末衍射图,图4所示为80℃干燥温度、干燥8小时的X-粉末衍射图,图5所示为60℃干燥温度、干燥10小时的X-粉末衍射图,图6所示为30℃干燥温度、干燥8小时的X-粉末衍射图。由此可知,虽然实施例1和实施例2中的水分子含量因真空干燥的温度和时间不同而不同,但是其X-粉末衍射的图基本一致,故本发明认为含有1~3摩尔水的(2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯并[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃-3-胺二盐酸盐都得到I型结晶。实施例3(2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃-3-胺-对甲苯磺酸盐(化合物2)(2R,3S,5R)-2-(2,5-difluorophenyl)-5-(1-methyl-2-(1-methyl-1H-tetrazol-5-yl)pyrrolo[3,4-d]imidazol-5(1H,4H,6H)-yl)tetrahydro-2H-pyran-3-amine4-methylbenzenesulfonate室温下,将1l(2.0g,4.8mmol)溶于乙酸乙酯(60mL)中,加入反应瓶,将对甲苯磺酸一水合物(0.912g,4.8mmol)溶于乙酸乙酯(20mL)中,滴加到上述反应液中,滴加完毕室温下反应2小时。反应液有白色固体析出,抽滤,滤饼用乙酸乙酯(10mL×3)、甲基叔丁基醚(10mL)洗涤,真空干燥,得到白色固体产物化合物2(2.5g,产率88.6%,HPLC纯度99.5%)。1HNMR(400MHz,DMSO-d6):δ8.01(s,3H),7.49(d,2H),7.38-7.33(m,3H),7.12(d,2H),4.55(d,1H),4.34(s,3H),4.21(m,1H),4.06-3.99(m,4H),3.87(m,2H),3.60(m,1H),3.41(m,2H),3.11(m,1H),2.50(m,1H),2.29(s,3H),1.72(m,1H)。实施例4(2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃-3-胺-二对甲苯磺酸盐(化合物3)(2R,3S,5R)-2-(2,5-difluorophenyl)-5-(1-methyl-2-(1-methyl-1H-tetrazol-5-yl)pyrrolo[3,4-d]imidazol-5(1H,4H,6H)-yl)tetrahydro-2H-pyran-3-aminebis(4-methylbenzenesulfonate)室温下,将1l(2.0g,4.8mmol)溶于乙酸乙酯(60mL)中,加入反应瓶,将对甲苯磺酸一水合物(1.824g,9.6mmol)溶于乙酸乙酯(20mL)中,滴加到上述反应液,滴加完毕室温下反应2小时。反应液有白色固体析出,抽滤,滤饼用乙酸乙酯(10mL×3)、甲基叔丁基醚(10mL)洗涤,真空干燥,得到白色固体产物化合物3(3.1g,产率85%,HPLC纯度99.63%)。1HNMR(400MHz,DMSO-d6):δ8.19(s,3H),7.49-7.47(m,4H),7.38-7.35(m,3H),7.11(d,4H),4.75-4.52(m,5H),4.41-4.34(m,4H),4.03(m,4H),3.67-3.62(m,2H),2.70(m,1H),2.27(s,6H),2.00(m,1H)。实施例5(2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃-3-胺-磷酸盐(化合物4)(2R,3S,5R)-2-(2,5-difluorophenyl)-5-(1-methyl-2-(1-methyl-1H-tetrazol-5-yl)pyrrolo[3,4-d]imidazol-5(1H,4H,6H)-yl)tetrahydro-2H-pyran-3-aminephosphate室温下,将1l(2.5g,6mmol)溶于乙酸乙酯(80mL)中,加入反应瓶,将磷酸(0.693g,6mmol,质量分数为85%)溶于乙酸乙酯(20mL)中,滴加到上述反应液,滴加完毕室温下反应2小时。反应液中有白色固体析出,抽滤,滤饼用乙酸乙酯(10mL×3)、甲基叔丁基醚(10mL)洗涤,真空干燥,得到白色固体产物化合物4(1.6g,产率52%,HPLC纯度99.53%)。1HNMR(400MHz,DMSO-d6):δ7.78(brs,5H),7.37-7.28(m,3H),4.50-4.37(m,1H),4.34(s,3H),4.19-4.16(m,1H),4.04-3.98(m,5H),3.86(m,2H),3.41-3.36(m,2H),3.10-3.05(m,1H),2.54(m,1H),1.70-1.67(m,1H)。实施例6(2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃-3-胺-苯甲酸盐(化合物5)(2R,3S,5R)-2-(2,5-difluorophenyl)-5-(1-methyl-2-(1-methyl-1H-tetrazol-5-yl)pyrrolo[3,4-d]imidazol-5(1H,4H,6H)-yl)tetrahydro-2H-pyran-3-aminebenzoate室温下,将1l(2.5g,6mmol)溶于乙酸乙酯(80mL)中,加入反应瓶,将苯甲酸(0.733g,6mmol)溶于乙酸乙酯(20mL)中,滴加到上述反应液,滴加完毕室温下反应2小时。反应液有白色固体析出,抽滤,滤饼用乙酸乙酯(10mL×3)、甲基叔丁基醚(10mL)洗涤,真空干燥,得到白色固体产物化合物5(2.1g,产率62%,HPLC纯度99.76%)。1HNMR(400MHz,DMSO-d6):δ7.95(d,2H),7.60(d,1H),7.49(t,2H),7.27-7.20(m,3H),4.34(s,3H),4.19-4.14(m,2H),4.03-3.98(m,5H),3.84(m,2H),3.30(t,1H),3.01-2.98(m,2H),2.35(d,1H),1.49-1.40(m,1H)。实施例7(2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃-3-胺-柠檬酸盐(化合物6)(2R,3S,5R)-2-(2,5-difluorophenyl)-5-(1-methyl-2-(1-methyl-1H-tetrazol-5-yl)pyrrolo[3,4-d]imidazol-5(1H,4H,6H)-yl)tetrahydro-2H-pyran-3-amine2-hydroxypropane-1,2,3-tricarboxylate室温下,将1l(2.5g,6mmol)溶于乙酸乙酯(80mL)中,加入反应瓶,将柠檬酸(1.152g,6mmol)溶于乙醇(20mL)中,滴加到上述反应液,滴加完毕室温下反应2小时。反应液中有白色固体析出,抽滤,滤饼用乙酸乙酯(10mL×3)、甲基叔丁基醚(10mL)洗涤,真空干燥,得到白色固体产物化合物6(3.2g,产率87.7%,HPLC纯度99.47%)。1HNMR(400MHz,MeOD):δ7.36-7.20(m,3H),4.67-4.65(m,1H),4.39-4.35(m,4H),4.15-4.09(m,2H),4.05-4.00(m,5H),3.59-3.54(m,2H),3.30-3.27(m,1H),2.86-2.72(m,4H),2.72-2.65(m,1H),1.87-1.81(m,1H)。实施例8(2R,3S,5R)-2-(2,5-二氟苯基)-5-(1-甲基-2-(1-甲基-1H-四唑-5-基)吡咯[3,4-d]咪唑-5(1H,4H,6H)-基)四氢-2H-吡喃-3-胺-富马酸盐(化合物7)(2R,3S,5R)-2-(2,5-difluorophenyl)-5-(1-methyl-2-(1-methyl-1H-tetrazol-5-yl)pyrrolo[3,4-d]imidazol-5(1H,4H,6H)-yl)tetrahydro-2H-pyran-3-aminefumarate室温下,将1l(2.5g,6mmol)溶于乙酸乙酯(80mL)中,加入反应瓶,将富马酸(0.696g,6mmol)溶于甲醇(20mL)中,滴加到上述反应液,滴加完毕室温下反应2小时。反应液中有白色固体析出,抽滤,滤饼用乙酸乙酯(10mL×3)、甲基叔丁基醚(10mL)洗涤,真空干燥,得到白色固体产物化合物7(2.3g,产率72%,HPLC纯度99.62%)。1HNMR(400MHz,DMSO-d6):δ7.31-7.23(m,3H),6.54(s,2H),4.34(m,4H),4.18-4.15(m,1H),3.99-3.98(m,5H),3.85(m,2H),3.33(t,1H),3.17-3.12(m,1H),3.05-3.00(m,1H),2.45(m,1H),1.60-1.52(m,1H)。实施例9化合物1重结晶将化合物1(53g,0.108mol)、去离子水(53mL)和无水乙醇(106mL)依次加入反应瓶中,加毕升温至80℃,待固体溶解完全,加入活性炭(5.3g),加毕,80℃下搅拌30分钟,趁热过滤,滤饼用热无水乙醇(26.5mL)洗涤。合并滤液,将醋酸异丙酯(344.5mL)滴加到滤液中,自然冷却至室温,析出白色固体,继续搅拌3小时,过滤,滤饼用醋酸异丙酯(60mL×2)洗涤,抽干,真空干燥(80℃,16小时),得到精制后的化合物1(45g,收率84.9%)。重结晶后化合物1晶型没有发生改变,重结晶前与重结晶后通过XRD图谱对比晶型一致。重结晶前XRD图谱为图8。重结晶前XRD图谱为图9。实施例10溶解度研究样品:化合物1(由表4中序号1条件得到)、化合物1l。实验:取2mL生理盐水和2mL0.5%CMC-Na溶液分别加入于BD离心管中,逐次加入样品,每次加入样品后在37℃水浴中加热并强力振摇或超声。待加入一定量样品后溶液变浑浊,在37℃水浴中加热30分钟并强力振摇或超声也不能使浑浊现象消失。实验结果见表5。表5溶解度结论:化合物1相对于化合物1l在生理盐水和0.5%CMC-Na溶液中的溶解度均显著提高。实施例11稳定性研究样品:化合物1l、化合物1、化合物2、化合物3、化合物4、化合物5、化合物6和化合物7。实验:取化合物1l、化合物1、化合物2、化合物3、化合物4、化合物5、化合物6、化合物7分别在高温(40℃和60℃)、高湿(RH75%和RH92.50%)、光照(5500±500lx)条件下进行试验,0天、10天、12天、14天、20天或30天的HPLC检测纯度(百分数表示),检测条件及实验结果见表6~9。HPLC检测纯度条件为:色谱柱:XselectCSHC18(4.6×150mm,3.5um);柱温:35℃;检测波长:275nm;流动相:按表3进行梯度洗脱;流速1.0mL/min;其中A相为10mmol/L磷酸氢二钾(pH=10.95),B相为甲醇。表6HPLC流动相T(min)0.014.0014.0020.0026.0030.0030.0137.00A(%)7070404010107070B(%)3030606090903030表7化合物1与化合物1l的稳定性对比表8化合物1的稳定性表9化合物2-7的稳定性结论:化合物1无论在高温、高湿还是光照条件下,10天后纯度基本没有变化,相比其游离碱化合物1l,稳定性明显提高。同时,化合物1在高温、高湿还是光照条件下,12天或30天后纯度基本没有变化,稳定性良好。化合物2、化合物3无论在高温、高湿还是光照条件下,14天或20天后纯度基本没有变化,稳定性良好。实施例12DPP-IV体外酶活测定利用重组人DPP-IV和H-Ala-Pro-AFC的酶学反应测定本发明化合物的DPP-IV体外酶活。按照DPP-IVFluorescentActivityAssayKit(BPSBioscience)配制缓冲液、待测样品工作液、DPP-IV酶稀释液和AFC底物稀释液。准备96孔板,每孔先加入80μL缓冲液,之后加入5μLDPP-AFC-底物。再加入不同浓度待测样工作液,每孔5μL,空白组加入5μL缓冲液。最后在测试组对照中加入10μLDPP-IV酶,空白对照组中加入10μL缓冲液。用Origin7.5软件对数据进行统计学分析,得到各测试化合物的IC50值,实验结果见表10。表10DPP-IV体外酶活测定结果序号化合物编号IC50(nM)1化合物11.372化合物22.023化合物32.114化合物42.225化合物51.806化合物61.577化合物71.90结论:本发明化合物具有明显的DPP-IV酶的抑制活性。当前第1页1 2 3
- 该技术已申请专利。仅供学习研究,如用于商业用途,请联系技术所有人。
- 技术研发人员:范江;陈清平;汪成涛
- 技术所有人:四川海思科制药有限公司
- 我是此专利的发明人
- 该领域下的技术专家
- 如您需求助技术专家,请点此查看客服电话进行咨询。
- 1、薛老师:1.CRISPR-Cas系统 2.基因编辑 3.基因修复 4.天然产物合成 5.单分子技术开发与应用
- 2、张老师:1.探索新型氧化还原酶结构-功能关系,电催化反应机制 2.酶电催化导向的酶分子改造 3.纳米材料、生物功能多肽对酶-电极体系的影响4. 生物电化学传感和生物电合成体系的设计与应用。
- 3、豆老师:1.环境纳米材料及挥发性有机化合物(VOCs) 2.CO污染物的催化氧化 3.低温等离子体 4.吸脱附等控制技术
- 4、赵老师:1.高分子材料改性及加工技术 2.微孔及过滤材料 3.环境友好高分子材料
- 5、邬老师:1.高分子材料的共混与复合 2.涉及材料功能化及结构与性能的研究; 高分子热稳定剂的研发
- 如您是高校老师,可以点此联系我们加入专家库。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
精彩留言,会给你点赞!