一种大量提取细菌质粒的方法与流程
本发明属于生物工程技术领域,具体涉及一种快速、简便、大量提取细菌质粒的方法。
背景技术:
质粒DNA的提取是分子生物学中基础工作之一,而获得足够用量的质粒DNA是保证后续转基因、BAC文库构建、细胞转染等研究工作的重要前提条件,只有在足够用量质粒基础上,才能进一步利用花粉管法或其他方法将外源基因导入动植物基因组中,并进而对特定基因功能进行研究。
现有技术中,为较为便捷的提取质粒,国内外很多生物试剂公司都研制了与提取质粒技术相关的商品化试剂盒。但该类试剂盒均以纯化柱为主,成本高,价格昂贵,并且利用其进行大量质粒DNA提取时,需要配置大量的培养液,并进行大量的摇菌培养,且需要特定的离心机,耗时费力,加大了实验的工作量,使得实验时间显著延长,影响生物学实验操作效率和实验结果。
就质粒提取方法而言,现有技术中,利用玻璃珠悬起菌落并快速提取质粒的方法在真核表达系统例如酵母菌株中有所应用,但该方法在原核表达系统尤其是大肠杆菌的质粒提取中是否能够获得较好的应用效果则缺乏相关的报道,同时由于真核表达系统与原核表达系统本身的巨大差异,该方法能否应用于原核表达系统也是未知的。由于原核表达系统应用十分广泛,因而极有必要就细菌中大量质粒提取方法进行进一步改进,从而为相关基因研究及原核表达系统的应用奠定良好基础。
技术实现要素:
本发明目的是提供一种简化的、较为快速的提取大量细菌质粒的方法,从而为相关基因研究奠定良好应用基础。
本发明所采取的技术方案详细介绍如下
一种大量提取细菌质粒的方法,包括如下步骤:
(1)筛选获得含有质粒的重组的原核细胞,具体为:
首先,在将目的质粒转化原核细胞后,挑取含有质粒的原核细胞的单菌落接种于含有抗性抗生素的液体培养基中,进行扩大培养;
其次,吸取扩大培养后的菌液涂布于含有抗性抗生素的固体培养基的平板上,培养过夜;
最后,在平板上加入无菌水和灭菌的玻璃珠摇晃,利用玻璃珠悬起菌落;待培养基中无菌落残留时,将含有菌落的无菌水吸出,置于离心管中,离心弃上清,保留沉淀,即为含有质粒的细菌细胞(原核细胞);
本申请中所述原核细胞为细菌细胞,具体例如为大肠杆菌细胞;
(2)裂解细胞,具体为:
在步骤(1)中离心后沉淀中加入溶液I,震荡重悬菌体,静置3~5min;然后加入溶液II ,混匀后,冰上放置3~5min;再加入溶液III,混合均匀,并冰上放置3~5min;
所述溶液I,每升溶液中的组份为:1M Tris-HCL(pH=8.0)、50mL,0.5M EDTA、20mL,其余为ddH2O,pH=7.5,高温灭菌;
所述溶液II,每升溶液的配制方法为:0.4M 的NaOH的ddH2O溶液与质量浓度2%的SDS的ddH2O溶液的等体积混合液;0.4M 的NaOH的ddH2O溶液可由8g NaOH 溶于500mL的ddH2O中配制而成,质量浓度2%的SDS的ddH2O溶液可由10g SDS溶于500mL的ddH2O中配制而成;需要强调的是,溶液II需现配现用;
所述溶液III,每升溶液的制备方法为:在600mL的ddH2O中依次加入129.69g的KAc和100mL冰乙酸,溶解并混合均匀后,乙酸调节pH=4.8,然后用ddH2O定容至1L,并最终调节pH=4.8;
(3)提取质粒,具体为:
将步骤(2)中冰上放置后溶液离心,弃沉淀,取上清液于离心管中,加入等体积量的氯仿-异戊醇混合液,混合均匀,放置15~25min;所述氯仿-异戊醇混合液中,以体积比计,氯仿:异戊醇=24:1;
将放置后混合液离心,取上清液置于预先加有无水乙醇的离心管中,离心,弃上清,再用体积分数75%的乙醇漂洗沉淀后,即为所提取的质粒;离心管中预先加入的无水乙醇的量优选为上清液2倍体积的量,所加入无水乙醇优选为-20℃预冷的无水乙醇;
所提取的质粒在干燥后,可溶于TE缓冲液中,-80℃保存备用。
本申请通过对细菌中质粒提取操作流程的优化,结果表明,利用玻璃珠悬起菌落并快速提取质粒的方法在原核表达系统中具有较好的应用效果,可较为快速的获得大量质粒,为转基因、BAC文库构建、细胞转染等应用研究奠定基础。由于该方法不需大量摇菌培养,操作较为方便,因而操作成本较为低廉;同时由于不需要大型的高速低温离心机的使用,且能够较为快速的大量提取质粒,因而对于缩短实验时间,提高实验效率具有较好地应用价值。
附图说明
图1为玻璃珠悬起菌落图;
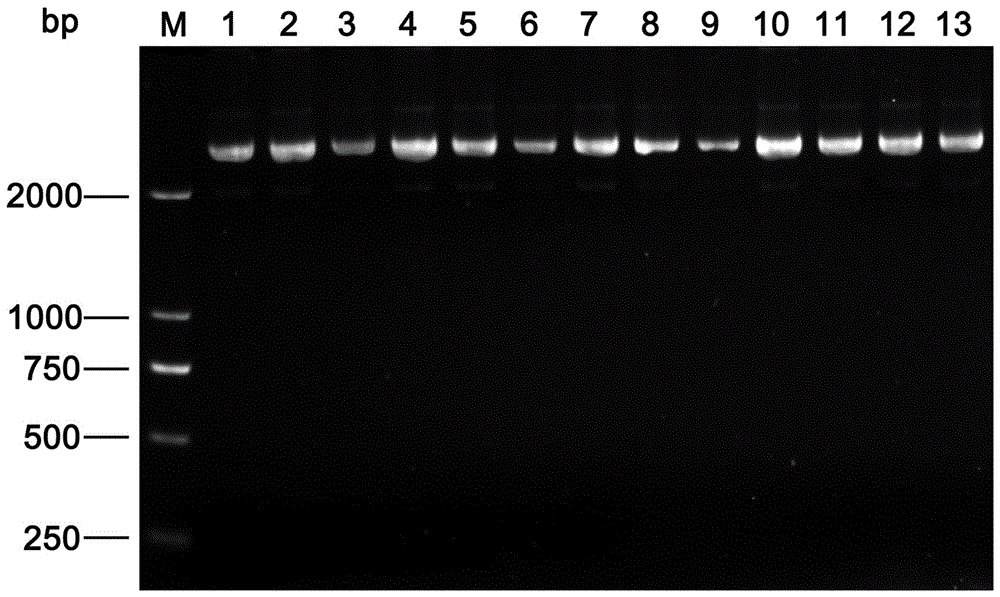
图2为本发明的质粒提取方法和常规大提质粒试剂盒的提取方法在提取重组质粒pMDC83- pTaWG04-GUS时的电泳结果对比图,其中:M为Marker Ⅳ,1-6泳道为挑取的6个阳性单克隆pMDC83-pTaWG04-GUS用常规试剂盒提取质粒的电泳图,7-13泳道为挑取的7个阳性单克隆pMDC83-pTaWG04-GUS用本发明的质粒提取方法提取质粒的电泳图;
图3为目标基因pTaWG04在提取质粒中的PCR扩增后的电泳图;其中M为Marker Ⅳ,1-3泳道为利用提取的质粒为模板进行目标条带pTaWG04的PCR扩增结果;
图4为质粒酶切结果;其中泳道M为Marker Ⅳ,泳道1为质粒小提酶切结果,泳道2为本发明方法所提质粒酶切结果。
具体实施方式
下面结合实施例对本发明做进一步的解释说明。介绍具体实施例前,对本发明中涉及的部分物料及试验设备情况简要介绍说明如下。
生物材料及实验试剂:
大肠杆菌DH5α菌株,为TaKaRa公司商品化产品;
植物表达载体pMDC83 :由华中农业大学作物遗传改良国家重点实验室涂金星教授赠予;
植物表达载体pBI101:购自于Clontech公司;
常规QIAGEN的大提质粒试剂盒:购自于QIAGEN公司;
常规小提质粒试剂盒:购自于天根生化科技公司;
溶液I,每升溶液中的组份为:1M Tris-HCL(PH=8.0)、50mL,0.5M EDTA、20mL,其余为ddH2O,PH=7.5,高温灭菌;
溶液II,每升溶液的配制方法为:0.4M 的NaOH的ddH2O溶液与质量浓度2%的SDS的ddH2O溶液的等体积混合液;0.4M 的NaOH的ddH2O溶液可由8g NaOH 溶于500mL的ddH2O中配制而成,2% SDS溶液可由10g SDS溶于500mL的ddH2O中配制而成;溶液II现配现用;
溶液III,每升溶液的制备方法为:在600mL的ddH2O中依次加入129.69g的KAc和100mL冰乙酸,溶解并混合均匀后,乙酸调节pH=4.8,然后用ddH2O定容至1L,最终确定pH=4.8;
溶液I、溶液II、溶液III配制时按现有技术配制即可,相关物料也为常规商品化试剂或物料,不再赘述。
实施例
本实施例以含有TaWG04启动子的pMDC83质粒为例,对该重组质粒进行了提取实验,相关过程简要介绍如下。
由于重组质粒的构建是相关转基因操作的基础,因而首先就含有TaWG04启动子(pTaWG04)的重组质粒的构建过程简要介绍如下, pTaWG04启动子碱基序列如SEQ ID NO.1所示。
第一,首先利用HindIII+EcoRI对pBI101进行双酶切,获得GUS基因,然后利用HindIII+EcoRI对质粒pMDC83进行双酶切,最后,利用T4 DNA连接酶将酶切产物进行连接,即,将GUS基因融合到pMDC83上,构建获得pMDC83-GUS重组载体。
酶切时的双酶切体系设计为:
10×Fast Digest buffer,6μL;
pMDC83质粒(或pBI101质粒),2μg、20μL;
HindIII,1U/L 、2μL;
EcoRI,1U/L 、2μL;
ddH2O,30 μL;
T4 DNA连接酶连接时的连接体系设计如下:
10×T4 DNA 连接缓冲液,1μL;
GUS基因片段,7μL;
pMDC83载体,1μL;
T4 DNA 连接酶,1μL。
第二,PCR扩增获得TaWG04启动子序列产物,具体步骤如下:
首先,设计引物,进行PCR扩增,获得TaWG04启动子序列;
引物序列设计如下(引物由上海生工合成提供):
pWG04-F:ACGCGTCGACACACAAGCTGCCTTTCCATAA;
pWG04-R:CCGCGGATCCTGAATTTGTGGTTACTCTTGAT;
10µL的PCR扩增体系设计如下:
5×phusion Buffer,2 μL;
dNTPmix,10mM、1.6 μL;
pGW04F,10µM、0.5 μL;
pGW04R,10µM、0.5 μL;
Phusion HS Ⅱ,2U/µL、0.2 μL;
TaWG04启动子DNA,10ng/µL、0.5 μL;
ddH2O,4.7 μL;
PCR反应(BIO-RAD S1000 Thermal Cycler PCR仪)程序:98℃,30sec;98℃、10sec,55℃、20sec,72℃、40sec,循环32次;72℃,10min;
对PCR反应产物进行1%琼脂糖凝胶电泳分离,EB染色。结果表明,所扩增目标条带长度为1557bp。进一步地,对目标条带进行切胶回收备用(采用上海生工质粒小提试剂盒进行操作)。
第三,将所获得TaWG04启动子序列产物连接到上述pMDC83-GUS重组载体上,具体步骤如下:
采用SalI和BamHI酶分别对所回收的TaWG04启动子扩增序列(第二步所制备)和pMDC83-GUS重组载体(第一步所制备)进行双酶切;回收酶切产物后,利用T4 DNA连接酶将酶切产物进行连接,即,将TaWG04启动子连接到pMDC83-GUS重组载体上,最终构建、筛选获得含有TaWG04启动子的重组质粒,命名为pMDC83-pTaWG04-GUS。
酶切时的双酶切体系设计为:
10×Fast Digest buffer,6μL;
pMDC83-GUS重组载体(或TaWG04启动子扩增产物),20μL;
SalI,1U/L 、2μL;
BamHI,1U/L 、2μL;
ddH2O,30 μL;
T4 DNA连接酶连接时的连接体系设计如下:
10×T4 DNA 连接缓冲液,1μL;
TaWG04启动子片段,7μL;
pMDC83-GUS载体,1μL;
T4 DNA 连接酶,1μL。
按现有常规操作,将含有TaWG04启动子的重组质粒pMDC83-pTaWG04-GUS转化大肠杆菌DH5α感受态细胞,LB培养基培养,并利用Kan(卡那霉素,浓度为0.1mg/mL)进行抗性筛选,对阳性菌株即可进行大量质粒提取操作。
利用本申请所提供的大量提取细菌质粒的方法进行质粒提取时,具体包括如下步骤:
(1)筛选获得含有质粒的原核细胞,具体为:
首先,在上述转化后、抗性筛选所获得菌落中,挑取阳性单菌落接种于2mL LB培养液中(培养液中含有Kan,浓度为0.1mg/mL),37℃培养3~4h后,于50mL的离心管中扩大培养4~6h;
其次,吸取上述扩大培养后的菌液50μL(或按4倍稀释成200μL)涂布于新的LB培养平板上(平板直径15cm,LB培养平板中含有Kan,浓度为0.1mg/mL),37℃过夜培养(12~16h);
最后,在上述长满菌落(过夜培养后,平板培养基中菌落基本可长满至整个平板)的培养基平板中加入4mL灭菌水,然后放入灭菌的玻璃珠轻轻摇晃(如图1所示),利用玻璃珠悬起菌落。待培养基中无菌落残留时,用移液枪(规格1000µL)将菌液(即含有菌落的无菌水)吸出,分别置于4个2mL的离心管中,12000 g离心5 min,弃去上清,保留沉淀,即为含有质粒的细菌细胞。
(2)裂解细胞,具体为:
在步骤(1)中离心后沉淀中(每个离心管中)加入600µL溶液I,在震荡器上剧烈震荡重悬菌体,静置5min;
然后加入600µL溶液II ,轻轻颠倒数次,彻底混匀(混匀过程中应确保离心管的整个内壁与溶液II 充分接触);混匀后(至溶液变清亮为止),冰上放置5min;
最后加入600µL溶液III,轻轻颠倒数次,混匀过程中看到絮状物时轻弹离心管使之均匀,同时,混匀过程中注意使溶液III在粘稠的细胞裂解物中分散均匀,混合均匀后冰上放置5min。
(3)提取质粒,具体为:
将步骤(2)中冰上放置后溶液4℃、12000 g离心10 min,弃沉淀,取上清液置于新的2mL离心管中;
加入等体积量的氯仿-异戊醇混合液,混合均匀,放置20min;所述氯仿-异戊醇混合液中,以体积比计,氯仿:异戊醇=24:1;
将放置后混合液4℃、12000 g离心10min,取上清液置于新的2mL离心管中(预先加入有2倍体积的-20℃预冷的无水乙醇),颠倒混匀,然后4℃、12000 g离心10min,弃上清;
用体积分数75%的乙醇漂洗沉淀两次后,即为所提取的质粒;
所提取的质粒在干燥后,可溶于1mL的 TE缓冲液中,取1µL作为样品,其余-80℃保存备用。
对本发明提取的质粒进行1%琼脂糖凝胶电泳分离,并EB染色,观察质粒片段大小及质量,结果如图2所示,图2中7-13泳道为随机所挑取的7个阳性单克隆pMDC83-pTaWG04-GUS的电泳图。
按照常规质粒提取试剂盒(QIAGEN)说明书,即,按照现有技术,对含有重组质粒的重组大肠杆菌进行质粒提取,以此作为对照对本申请技术效果进行展示,提取时具体包括如下步骤:
(1)把含有重组载体pMDC83-pTaWG04-GUS的菌液取500μl加到250mL LB中,37℃过夜摇菌,然后用250mL离心瓶4000rpm离心15min收集菌体;
(2)去除上清后,加入4mL P1重悬,转移到50mL离心管中;
(3)加入4mL P2,上下颠倒至混合物完全变蓝,室温放置5min;
(4)加入4mL P3上下颠倒至混合物完全变成白色,置于冰上放置15min,20000g、4℃离心30min,吸取上清到新的50mL离心管中,20000g、4℃离心15min;
(5)离心时吸取4mL QBT加到QIAGEN-Tip100柱子中使其自然流下,将离心结束后50mL离心管中的上清转移到QIAGEN-Tip100柱子中,使其自然流下;
(6)待液体流尽,用10mL QC漂洗QIAGEN-Tip100柱子两次,最后用5mL的QF将质粒洗脱;
(7)在洗脱的质粒溶液中加入3.5mL异丙醇沉淀质粒DNA,17000g、4℃离心5min收集质粒,将收集的质粒晾干,加入适合体积的ddH2O溶解质粒。
对常规试剂盒提取的质粒进行1%琼脂糖凝胶电泳分离,并EB染色,观察质粒片段大小及质量,结果如图2所示,图2中1-6泳道为随机挑取的6个阳性单克隆的电泳图。
对比两种质粒提取的效果发现,本发明的提取方法在质粒提取效果上跟常规商品化的质粒提取试剂盒(QIAGEN)基本一致。具体而言:
在6个常规质粒试剂盒的提取结果和7个本发明的质粒提取结果的对比中可以发现,本发明在提取效果上更稳定,如泳道7、10、11、12、13的条带亮度基本一致,而常规商品化的质粒提取试剂盒只有泳道2、4的条带亮度和本发明的一致。推测这种结果可能是由于商品化提取操作过程过于繁琐所造成的,或者说,这种商品化的质粒提取试剂盒对实验人员的操作技能的要求更加严格。
而基于上述对比结果,可以看出,本发明不需大量摇菌培养,操作较为方便,操作成本较为低廉;同时由于不需要大型的高速低温离心机的使用,且能够较为快速的大量提取质粒,因而对于缩短实验时间,提高实验效率具有较好地应用价值。
检验例
对于上述所提取重组后的pMDC83-pTaWG04-GUS质粒(本发明方法中所提取),发明人进一步进行了PCR检测和双酶切检测,以检验所提取质粒是否正确。具体检测过程简要介绍如下。
检测
对所提取的含有TaWG04的pMDC83重组质粒进行PCR扩增检测验证时,所用PCR扩增引物同前述引物序列(pGW04F、pGW04R),10µL的PCR扩增体系设计如下:
10×Buffer,1 μL;
dNTPmix,10mM、0.8 μL;
pGW04F,10µM、0.4μL;
pGW04R,10µM、0.4 μL;
rTaq DNA聚合酶,0.5U/μL、0.1 μL;
pMDC83-pTaWG04-GUS重组质粒,10ng/µL、0.5 μL;
ddH2O,6.8 μL;
PCR反应程序:94℃,5min;94℃、1min,55℃、1min,72℃、1min,循环30次;72℃,5min。
对PCR反应产物进行凝胶电泳。结果如图3所示,从图中可以看出,扩增出目标条带大小约为1500bp,与预期的TaWG04启动子长度相同,表明所提取质粒中成功整合含有TaWG04启动子序列。
双酶切检测
对所提取质粒样品采用SalI和BamHI进行双酶切检测,20µL酶切体系设计如下:
10 ×FastDigest buffer,2 μL;
SalI,1U/L、1 μL;
BamHI,1U/L、1 μL;
质粒DNA,1 μg;
ddH2O,加至20µL;
37℃酶切30min。
酶切产物进行1%琼脂糖凝胶电泳分离,EB染色,观察酶切片段大小。
按照小提质粒提取试剂盒说明书,即,按照现有技术,提取少量质粒,对所提取质粒进行双酶切,以此作为对照,以对本申请中所提取质粒的酶切效果进行展示。提取质粒过程具体如下:
(1)吸取2mL经过37℃充分震荡培养16h的菌液pMDC83-pTaWG04-GUS,8000g离心2min,倒掉液体;
(2)向菌体中加入250μL Buffer P1,使菌体混匀悬浮;
(3)吸取Buffer P2 250μL加入到离心管中,轻轻晃动离心管混匀,然后放置10min;
(4)吸取Buffer P3 350μL ,轻轻晃动离心管,放置10min,12000g离心8min;
(5)取吸附柱将上清全部移入,8000g离心2min,倒出废液;
(6)吸取Wash Solution 500μL加入到吸附柱中,8000g离心2min,倒出废液,并重复步骤一次;
(7)将空吸附柱和收集管放入小型离心机中,9000g离心1min;
(8)在吸附膜中央加入30μL Elution Buffer,室温静置5min,9000g离心1min,此时离心管底部的溶液就是提取的质粒。
参考上述酶切体系对采用现有方法所提取质粒进行双酶切,并进行电泳检测。图4中,泳道M为Marker,泳道1为采用现有小量质粒提取试剂盒所提取质粒的酶切结果,泳道2为本方法所提取的质粒酶切结果。从图中可以看出,本发明所提取质粒的条带相对更为明亮,即,酶切效果更佳。
从上述本发明与现有大、小提商品化提取质粒试剂盒的效果对比可以看出,本发明所提取质粒条带更为明亮、质粒含量多、提取效果较好,加上其不需大量摇菌培养、操作方便、成本较为低廉、对仪器设备要求简单,如不需大型的高速低温离心机的使用、且能够较为快速的大量提取质粒,因而对于缩短实验时间,提高实验效率具有较好地应用价值。
SEQUENCE LISTING
<110> 河南省农业科学院小麦研究所
<120> 一种大量提取细菌质粒的方法
<130> none
<160> 1
<170> PatentIn version 3.5
<210> 1
<211> 1571
<212> DNA
<213> Triticum aestivum
<400> 1
tagatcccct gttgtaattt atcaaaaatt tatacacaag ctgcctttcc ataatttgaa 60
acattgtact ttctacttgc ctgcaggaag aactcttttg ttatggaccc ccttatctgg 120
cgaatagtcg ccaggagggg gtggcatttg cgaatgattc gctggcagtt ttagtttgag 180
ggatggaagt ttacatcagg gcgacatgac aatttctttt tgagaaggca aattgttgtt 240
ttaaataggc atcgtttgat cttgtctcgg aaccgaaact gccatcgaaa gagtttcgtt 300
tgccacctct ctcccagcaa acgattgcca attatgatta ccgtttgtta tatgcatgaa 360
ctgtgcgtct acagttggcg gcagcgaggt tgaggagacc ggtgtcatcg gcagcggctt 420
cagccggtgg aggcgtccta tggttgacgg ccgtgctcct tcctgctcga gaagagaagt 480
gagagggtgg tgagaggtga agagagcagg aggcggcagc ctggtgctgc tgcgggtcgt 540
agtaggagcc aaggcggcgg tgctgggtgg ctcgggctgt tatgcgggcg cgcgagcgac 600
accaagccct caacaaccgt gtccgtttca gttgggtctt ccatgttggg tacactggtg 660
gtgtccggct ctccgtctgc gtccattgtc cgttttggcg acccaaacgg acaaaaagcg 720
gacaaaaatt cgtgtccgtt tgagtcattg cgttgaagtt ggccttatgc tattccacag 780
tatactcaac gcactctaat tcacattgca tgctattaca tgcgtcattg tatttgatac 840
accgtgtgca atattttttt tctggtgaca atatatctac tatcgaacca agtataaagt 900
tcctcaaagg gtttaatatc atgttcatgg tgttcccgcg gaaatgcatg ggaatcatct 960
agtgtctatt aacaactata cgacattact aacattggca taaaaaaaag aattgatgag 1020
tcatgtatgc ttgttcatct taccatcata ttacacaaaa ctacgaagtt agttcagaaa 1080
gaaatagtct agaacaatct tcatattaat agtatgatct atcttaacaa cgccaagcaa 1140
gactataaac ttagttcccg acaagctatg ccaacctaga tgtgcctaac aacttgcaga 1200
acattacaaa cttagtttta gaaaggggtg taatatagat aagtgtttcg catgtaaaat 1260
gaatatgatg agtcattacg attatcaagc tttcctggca ctccatggat gtgtgctgca 1320
agaaagcaac tttggcgatc aatctgaaaa ttacacttgt aagtagcgcc accgaacaaa 1380
acataccaaa ggatcagttt gataagagta gaggaacttt acaagaaagc aaatgtgaag 1440
acgaaaagaa atcatttcat ggcagctata aatagccata cgccatgaag acccccttcc 1500
atcatccatc cttcagaaat ttggagcaca agcatccata ataaacaatc aagagtaacc 1560
acaaattcac c 1571
- 还没有人留言评论。精彩留言会获得点赞!