一种斑蝥素的新型绿色环保合成工艺的制作方法
本发明涉及一种斑蝥素的制备方法,具体涉及在常压条件下制备斑蝥素的绿色环保合成工艺,属于药物合成与制备领域。
背景技术:
斑蝥素主要来源于南方大斑蝥或黄黑小斑蝥的干燥体。药理研究表明斑蝥具有抗肿瘤活性,对肝癌、宫颈癌、皮肤癌、骨髓癌、白血病等均有抑制作用。斑蝥素对癌症患者正常细胞的损伤较小,同时斑蝥素有升高白细胞,不抑制免疫系统等优点,因此,斑蝥素一直都是抗肿瘤药物的研发热点。由于天然资源的匮乏,斑蝥素、斑蝥酸钠和甲基斑蝥胺等片剂和注射液无法大量的生产。
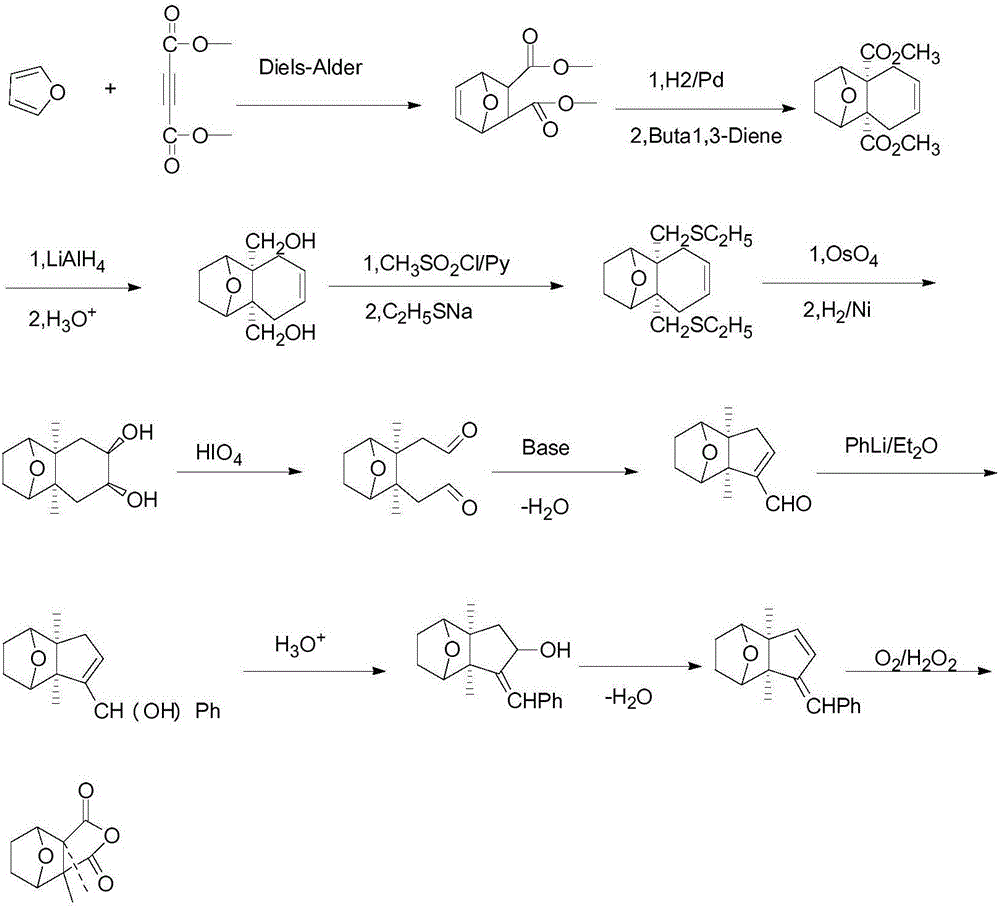
自1914年Gadamer证实了斑蝥素的结构(图1)以后,各国药物合成学家一直致力于斑螯素的化学合成,1951年Stork首次经过11步(图2)全合成了斑蝥素,1953年Schenk经过7步合成(图3),1976年Dauben经过2步合成(图4)。虽然他们都可以合成得到斑蝥素,但是在制备很多中间体时反应条件十分的苛刻,具体操作比较麻烦,斑蝥素的收率比较低。特别是Dauben的反应需要在15000个大气压条件下进行,这对设备要求十分高,很难实现工业化生产。
一直以来,如何降低制备斑螯素关键环加成反应时的苛刻条件、同时提高斑蝥素的收率是各国科研人员的研发热点。1990年Grieco等在研究环加成反应时候提出用高氯酸锂来缩短反应时间以及提高目标物的收率,但高氯酸锂对反应的无水条件要求很严格,操作很困难。因此需要开发一种反应条件温和,且斑蝥素收率高的新工艺来制备斑螯素。
技术实现要素:
针对现有合成斑蝥素环加成反应条件十分苛刻的问题,如需要超高压,且转化率低等问题,本发明提出了一种常压下斑蝥素的新型绿色环保合成工艺。
为实现上述目的,本发明采用如下技术方案:
(1)制备3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩
将NaCN水溶液加入反应釜中,冰浴降温,快速加入亚硫酸氢钠水溶液调节反应液的PH为7~8,然后慢慢滴加3-氧代-4-甲酸甲酯噻吩的甲醇溶液,3-氧代-4-甲酸甲酯噻吩与NaCN的质量比为1.8~3.3∶1,滴毕后升至室温反应10~18h,然后用二氯甲烷萃取三次,真空下蒸发掉二氯甲烷,得黄色油状物,即3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩;
(2)制备3-氰基-4-甲酸甲酯-2,5-二氢噻吩
冰浴条件下,将步骤(1)制得的3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩加入反应器中,加入苯与吡啶,搅拌均匀,控制内温在0~10℃,慢慢滴加POCl3,其中3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩∶苯∶吡啶∶三氯氧磷的质量比为1∶(0.88~1.32)∶(1.5~2)∶(0.8~4.10),反应液由淡黄色变为红棕色,最后变为褐色,滴毕后升至室温反应1~4h,再升温至50~60℃反应0.5~2h;二氯甲烷萃取,硅胶柱分离得3-氰基-4-甲酸甲酯-2,5-二氢噻吩;将剩余POCl3用饱和的碳酸氢钠溶液处理;
(3)制备2,5-二氢噻吩-3,4-二羧酸
将3-氰基-4-甲酸甲酯-2,5-二氢噻吩、乙酸、浓盐酸加入反应釜中回流反应,3-氰基-4-甲酸甲酯-2,5-二氢噻吩∶乙酸∶浓盐酸的固液比为1g∶(1.35~4.05)mL∶(3.5~6.76)mL,TLC监测反应完全以后,蒸干用丙酮提取得到黄色2,5-二氢噻吩-3,4-二羧酸粉末;
(4)制备2,5-二氢噻吩-3,4-二羧酸酐
将2,5-二氢噻吩-3,4-二羧酸与二氯亚砜加入反应釜中搅拌回流,2,5-二氢噻吩-3,4-二羧酸∶二氯亚砜的质量比为1∶2~8.0;反应完全后,加热蒸干,用二氯甲烷重结晶得黑色针状晶体,用乙腈重结晶得黄白色针状晶体:2,5-二氢噻吩-3,4-二羧酸酐;
(5)制备斑蝥素
按2,5-二氢噻吩-3,4-二羧酸酐∶呋喃∶离子液体的固液比为=1mg∶(3.5~6)μl∶(2~4)μl,加入反应器中,加热至20~50℃搅拌反应18~25小时,降至室温,用乙酸乙酯萃取,HPLC测试纯度。加入催化量的Raney-Ni回流反应3~5h,抽滤,乙酸乙酯洗涤,硅胶柱分离,得白色晶体,即斑蝥素。
进一步的,在步骤(4)的反应釜中加入苯作为有机溶剂溶解2,5-二氢噻吩-3,4-二羧酸,以提高产物2,5-二氢噻吩-3,4-二羧酸酐的收率。具体步骤为:在反应釜中加入2,5-二氢噻吩-3,4-二羧酸与苯搅拌均匀,待2,5-二氢噻吩-3,4-二羧酸溶解完全后,再加入二氯亚砜,2,5-二氢噻吩-3,4-二羧酸∶二氯亚砜的质量比为1∶2~8.0;反应完全后,加热蒸干,用二氯甲烷重结晶得黑色针状晶体,用乙腈重结晶得黄白色针状晶体:2,5-二氢噻吩-3,4-二羧酸酐;
进一步的,在步骤(5)中加入催化量的三氟化硼乙醚作为催化剂,加快反应进程。
进一步的,所述离子液体优选咪唑类离子液体。
使用本发明所述的方法合成斑蝥素中间体,不需要超高压的条件和设备,在20~50℃过夜反应即可,条件十分温和,转化率高,且斑蝥素收率高,为斑蝥素的工业化生产提供了十分有利的条件。
附图说明
图1是斑蝥素的结构。
图2是传统斑蝥素合成路线(11步)。
图3是传统斑蝥素合成路线(7步)。
图4是传统斑蝥素合成路线(2步)。
图5是本发明斑蝥素合成路线。
具体实施方式
下面结合附图对本发明的实施方式进行详细说明。以下实施例中使用的浓盐酸均为质量分数为37%的浓盐酸。
实施例1
(1)制备3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩
冰浴条件下将315g NaCN水溶液(质量百分比浓度为23.2%)、620mL纯化水,加入5L反应釜中,快速加入亚硫酸氢钠水溶液调节反应液的PH为7~8,40min内慢慢滴加132g/75mL的3-氧代-4-甲酸甲酯噻吩/甲醇溶液,滴毕后升至室温反应16h。用二氯甲烷萃取三次,真空下蒸发掉二氯甲烷,得197g黄色油状物3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩,收率80%。
(2)制备3-氰基-4-甲酸甲酯-2,5-二氢噻吩
将40g 3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩、35.2g苯、60g吡啶,加入1L的反应釜中,控制内温在0~3℃,慢慢滴加164g POCl3,待反应液由淡黄色变为红棕色,最后变为褐色,滴毕后升至室温反应4h,升温至50~52℃反应1h。二氯甲烷萃取,硅胶柱分离得3-氰基-4-甲酸甲酯-2,5-二氢噻吩,剩余POCl3用饱和的碳酸氢钠溶液处理。ESI-MS:[M+1]+170.02;1H NMR(CCl3D,TMS)δ:3.86(s,3H),4.04~4.06(m,2H),4.08~4.10(m,2H);13C NMR(CCl3D)δ:38.3,40.3,52.9,113.3,120.5,146.6,161.4。
(3)制备2,5-二氢噻吩-3,4-二羧酸
将3.6g 3-氰基-4-甲酸甲酯-2,5-二氢噻吩、11mL乙酸、19mL浓盐酸加入反应釜中,回流反应,TLC监测反应完全以后,蒸干反应液,用丙酮提取得到3.5g黄色2,5-二氢噻吩-3,4-二羧酸粉末,2,5-二氢噻吩-3,4-二羧酸的收率为94%。ESI-MS[M+1]+:175.02;1H NMR(DMSO)δ:3.96(s,4H);13C NMR(DMSO)δ:40.0,138.5,165.7。
(4)制备2,5-二氢噻吩-3,4-二羧酸酐
将0.5g 2,5-二氢噻吩-3,4-二羧酸、4g二氯亚砜加入反应釜中搅拌回流反应,待反应液停止产生气泡为反应完全,加热蒸干反应液,依次用二氯甲烷、乙腈重结晶得黄白色针状晶体2,5-二氢噻吩-3,4-二羧酸酐0.2g,收率为44%。ESI-MS[M+23]+:178.98;1H NMR(CCl3D,TMS)δ:4.00(s,4H);13C NMR(CCl3D)δ:31.5,153.9,158.5。
(5)制备斑蝥素
向1mL反应小瓶中,加入26mg 2,5-二氢噻吩-3,4-二羧酸酐、91微升呋喃、52微升1-丁基-3-甲基咪唑四氟硼酸盐,加热至50℃搅拌反应19小时,降至室温,3*1mL乙酸乙酯萃取,目标物HPLC 95%(2,5-二氢噻吩-3,4-二羧酸酐含量为0)。加入催化量的Raney-Ni回流反应4h,HPLC检测斑蝥素含量92%,抽滤,3*1mL乙酸乙酯洗涤,硅胶柱分离得30.8mg白色晶体,斑蝥素收率90%。
ESI-MS[M+23]+:219.35;1H NMR(CCl3D,TMS)δ:1.23(s,6Η),1.76(m,4Η),4.71(d,2Η);13C NMR(CCl3D)δ:12.7,23.4,55.2,84.7,175.9。
实施例2
(1)制备3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩
冰浴条件下将420g NaCN水溶液(质量23.2%)、800mL纯化水,加入5L反应釜中,快速加入亚硫酸氢钠水溶液调节反应液的PH为7~8,慢慢滴加244g/100mL的3-氧代-4-甲酸甲酯-噻吩/甲醇溶液,滴毕后反应16h。二氯甲烷萃取三次,真空下蒸发掉二氯甲烷,得250g黄色油状物3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩,收率76%。
(2)制备3-氰基-4-甲酸甲酯-2,5-二氢噻吩
控制内温在4~6℃,将80g 3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩、88g苯、140g吡啶投入2L反应釜中,慢慢滴加250g POCl3,待反应液由淡黄色变为红棕色,最后变为褐色,滴毕后升室温反应1h,升温55~60℃反应2h。二氯甲烷萃取,硅胶柱分离得3-氰基-4-甲酸甲酯-2,5-二氢噻吩,将剩余POCl3用饱和的碳酸氢钠溶液处理。ESI-MS:[M+1]+170.02;1H NMR(CCl3D,TMS)δ:3.86(s,3H),4.04~4.06(m,2H),4.08~4.10(m,2H);13C NMR(CCl3D)δ:38.3,40.3,52.9,113.3,120.5,146.6,161.4。
(3)制备2,5-二氢噻吩-3,4-二羧酸
将7.6g 3-氰基-4-甲酸甲酯-2,5-二氢噻吩、22mL乙酸、36mL浓盐酸加入反应釜中,回流反应,TLC监测反应完全以后,蒸干反应液,用丙酮提取得到6.5g黄色粉末,收率83%。ESI-MS[M+1]+:175.02;1H NMR(DMSO)δ:3.96(s,4H);13C NMR(DMSO)δ:40.0,138.5,165.7。
(4)制备2,5-二氢噻吩-3,4-二羧酸酐
将2.5g 2,5-二氢噻吩-3,4-二羧酸与16.4g二氯亚砜加入反应釜中搅拌回流反应,待反应液停止产生气泡为反应完全,加热蒸干反应液,依次用二氯甲烷、乙腈重结晶得黄白色针状晶体1g,收率为44%。ESI-MS[M+23]+:178.98;1H NMR(CCl3D,TMS)δ:4.00(s,4H);13C NMR(CCl3D)δ:31.5,153.9,158.5。
(5)制备斑蝥素
52mg 2,5-二氢噻吩-3,4-二羧酸酐、250微升呋喃、130微升1-丁基-3-甲基咪唑二(三氟甲基磺酰亚胺),加入反应瓶中,加热至40℃反应25小时,降至室温,3*1mL乙酸乙酯萃取,溶液呈浅黄色,HPLC检测:目标物含量94%,2,5-二氢噻吩-3,4-二羧酸酐0%。加入催化量的Raney-Ni回流反应3h,HPLC检测:斑蝥素含量90%,抽滤,3*1mL乙酸乙酯洗涤,硅胶柱分离得48.5mg白色晶体,斑蝥素收率80%。ESI-MS[M+23]+:219.35;1H NMR(CCl3D,TMS)δ:1.23(s,6Η),1.76(m,4Η),4.71(d,2Η);13C NMR(CCl3D)δ:12.7,23.4,55.2,84.7,175.9。
实施例3
(1)制备3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩
冰浴条件下将500g NaCN水溶液(质量23.2%)、1000mL纯化水,加入10L反应釜中,快速加入亚硫酸氢钠水溶液调节反应液的PH为7~8,慢慢滴加336g/120mL的3-氧代-4-甲酸甲酯-噻吩/甲醇溶液,滴毕后反应16h。二氯甲烷萃取三次,真空下蒸发掉二氯甲烷,得295g黄色油状物3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩,收率75%。
(2)制备3-氰基-4-甲酸甲酯-2,5-二氢噻吩
控制内温在8~10℃,将160g 3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩、176g苯、280g吡啶加入2L反应釜中,慢慢滴加500g POCl3,待反应液由淡黄色变为红棕色,最后变为褐色,滴毕后升室温反应2h,升温53~55℃反应0.5h。二氯甲烷萃取,过柱得3-氰基-4-甲酸甲酯-2,5-二氢噻吩;将剩余POCl3用饱和的碳酸氢钠溶液处理。ESI-MS:[M+1]+170.02;1H NMR(CCl3D,TMS)δ:3.86(s,3H),4.04~4.06(m,2H),4.08~4.10(m,2H);13C NMR(CCl3D)δ:38.3,40.3,52.9,113.3,120.5,146.6,161.4。
(3)制备2,5-二氢噻吩-3,4-二羧酸
将36g 3-氰基-4-甲酸甲酯-2,5-二氢噻吩、110mL乙酸、180mL浓盐酸加入反应釜中,回流反应,TLC监测反应完全以后,蒸干反应液,用丙酮提取得到30g黄色粉末,收率80%。ESI-MS[M+1]+:175.02;1H NMR(DMSO)δ:3.96(s,4H);13C NMR(DMSO)δ:40.0,138.5,165.7。
(4)制备2,5-二氢噻吩-3,4-二羧酸酐
将16g 2,5-二氢噻吩-3,4-二羧酸与87.5g二氯亚砜加入反应釜中搅拌回流反应,待反应液停止产生气泡为反应完全,加热蒸干反应液,依次用二氯甲烷重、乙腈重结晶得黄白色针状晶体2,5-二氢噻吩-3,4-二羧酸酐6g,收率为33%。ESI-MS[M+23]+:178.98;1H NMR(CCl3D,TMS)δ:4.00(s,4H);13C NMR(CCl3D)δ:31.5,153.9,158.5。
(5)制备斑蝥素
130mg 2,5-二氢噻吩-3,4-二羧酸酐、650微升呋喃、390微升1-丁基-3-甲基咪唑六氟磷酸盐、1滴三氟化硼乙醚,加入反应瓶中,加热至35℃反应18小时,降至室温,3*1mL乙酸乙酯萃取,溶液呈浅黄色,HPLC检测:目标物含量94%,酸酐0.4%。加入催化量的Raney-Ni回流反应5h,HPLC检测:斑蝥素含量90%,抽滤,3*1mL乙酸乙酯洗涤,旋除部分溶剂,硅胶柱分离得110mg白色晶体,斑蝥素收率76%。ESI-MS[M+23]+:219.35;1H NMR(CCl3D,TMS)δ:1.23(s,6Η),1.76(m,4Η),4.71(d,2Η);13C NMR(CCl3D)δ:12.7,23.4,55.2,84.7,175.9。
实施例4
(1)制备3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩
将629g NaCN水溶液(质量23.2%)、1250mL水溶液加入10L反应釜中,冰浴降温,快速加入亚硫酸氢钠水溶液调节反应液的PH为7~8,在40min内滴加400g/150mL的3-氧代-4-甲酸甲酯-噻吩/甲醇溶液,滴毕后升至室温反应18h。用二氯甲烷萃取三次,真空下蒸发掉二氯甲烷旋干,得360g黄色油状物,收率为73%。
(2)制备3-氰基-4-甲酸甲酯-2,5-二氢噻吩
控制内温在5~7℃,将200g 3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩、220g苯、350g吡啶加入5L反应釜中,慢慢滴加160g POCl3,待反应液由淡黄色变为红棕色,最后变为褐色,滴毕后升至室温反应2h,升温至53~54℃反应1h。二氯甲烷萃取,硅胶柱分离得3-氰基-4-甲酸甲酯-2,5-二氢噻吩。ESI-MS:[M+1]+170.02;1H NMR(CCl3D,TMS)δ:3.86(s,3H),4.04~4.06(m,2H),4.08~4.10(m,2H);13C NMR(CCl3D)δ:38.3,40.3,52.9,113.3,120.5,146.6,161.4。
(3)制备2,5-二氢噻吩-3,4-二羧酸
将72g 3-氰基-4-甲酸甲酯-2,5-二氢噻吩、200mL乙酸、250mL浓盐酸加入反应釜中,回流反应,TLC监测反应完全以后,蒸干反应液,用丙酮提取得到44.7g黄棕色粉末,收率87%。ESI-MS[M+1]+:175.02;1H NMR(DMSO)δ:3.96(s,4H);13C NMR(DMSO)δ:40.0,138.5,165.7。
(4)制备2,5-二氢噻吩-3,4-二羧酸酐
将36g 2,5-二氢噻吩-3,4-二羧酸与131g二氯亚砜、50mL苯加入反应釜中搅拌,待反应液停止产生气泡为反应完全,加热蒸干反应液,依次用二氯甲烷、乙腈重结晶得黄白色针状晶体25g,收率为78%。ESI-MS[M+23]+:178.98;1H NMR(CCl3D,TMS)δ:4.00(s,4H);13C NMR(CCl3D)δ:31.5,153.9,158.5。
(5)制备斑蝥素
260mg 2,5-二氢噻吩-3,4-二羧酸酐、1430微升呋喃、910微升1-已基-3-甲基咪唑四氟硼酸盐、1滴三氟化硼乙醚,加入反应瓶中,加热32~34℃搅拌反应20小时,降至室温,3*1mL乙酸乙酯萃取,溶液呈浅黄色,HPLC检测:目标物含量92%,酸酐0.77%。加入催化量的Raney-Ni回流反应4h,HPLC检测:斑蝥素含量85%,抽滤,3*1mL乙酸乙酯洗涤,旋除部分溶剂,过硅胶柱分离得212mg白色晶体,斑蝥素收率为74%。ESI-MS[M+23]+:219.35;1H NMR(CCl3D,TMS)δ:1.23(s,6Η),1.76(m,4Η),4.71(d,2Η);13C NMR(CCl3D)δ:12.7,23.4,55.2,84.7,175.9。
实施例5
(1)制备3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩
冰浴条件下将965g NaCN水溶液(质量23.2%)、2500mL纯化水加入10L反应釜中,快速加入亚硫酸氢钠水溶液调节反应液的PH为7~8。然后在40min内滴加840g/300mL的3-氧代-4-甲酸甲酯-噻吩/甲醇溶液,滴毕后升至室温反应10h。用二氯甲烷萃取三次,真空下蒸发掉二氯甲烷旋干,得690g黄色油状物,收率为70%。
(2)制备3-氰基-4-甲酸甲酯-2,5-二氢噻吩
将600g 3-氰基-3-羟基-4-甲酸甲酯-2,5-四氢噻吩加入5L的反应瓶中,加入792gmL苯与1200g吡啶,搅拌均匀,控制内温在0~2℃,慢慢滴加1580g POCl3,反应液由淡黄色变为红棕色,最后变为褐色,滴毕后升至室温反应2h,升温至57℃反应1h。将剩余POCl3用饱和的碳酸氢钠溶液处理,二氯甲烷萃取,过柱得3-氰基-4-甲酸甲酯-2,5-二氢噻吩。ESI-MS:[M+1]+170.02;1H NMR(CCl3D,TMS)δ:3.86(s,3H),4.04~4.06(m,2H),4.08~4.10(m,2H);13C NMR(CCl3D)δ:38.3,40.3,52.9,113.3,120.5,146.6,161.4。
(3)制备2,5-二氢噻吩-3,4-二羧酸
将144g 3-氰基-4-甲酸甲酯-2,5-二氢噻吩、583mL乙酸、973mL浓盐酸加入反应釜中,回流反应,TLC监测反应完全以后,蒸干反应液,用丙酮提取得到87.5g黄棕色粉末,收率85%。ESI-MS[M+1]+:175.02;1H NMR(DMSO)δ:3.96(s,4H);13C NMR(DMSO)δ:40.0,138.5,165.7。
(4)制备2,5-二氢噻吩-3,4-二羧酸酐
将72g 2,5-二氢噻吩-3,4-二羧酸与144g二氯亚砜、100mL苯加入反应釜中搅拌回流反应,待反应液停止产生气泡为反应完全,加热蒸干反应液,依次用二氯甲烷、乙腈重结晶得黄白色针状晶体50g,收率为78%。ESI-MS[M+23]+:178.98;1H NMR(CCl3D,TMS)δ:4.00(s,4H);13C NMR(CCl3D)δ:31.5,153.9,158.5。
(5)制备斑蝥素
520mg 2,5-二氢噻吩-3,4-二羧酸酐、呋喃3120微升、1-辛基-3甲基咪唑四氟硼酸盐2080微升,加入反应瓶中,加热20~25℃反应25小时,降室温,3*1mL乙酸乙酯萃取,溶液呈浅黄色,HPLC检测:目标物含量90.26%,酸酐0.9%。加入催化量的Raney-Ni回流反应4h,HPLC检测:斑蝥素含量83%,抽滤,3*1mL乙酸乙酯洗涤,旋除部分溶剂,硅胶柱分离得400mg白色晶体,斑蝥素收率66%。
ESI-MS[M+23]+:219.35;
1H NMR(CCl3D,TMS)δ:1.23(s,6Η),1.76(m,4Η),4.71(d,2Η);13C NMR(CCl3D)δ:12.7,23.4,55.2,84.7,175.9。
- 还没有人留言评论。精彩留言会获得点赞!