基于重组酶和超保真DNA聚合酶的大载体DNA的定点突变方法与流程
本发明属于生物技术领域,具体涉及一种基于重组酶和超保真dna聚合酶的大载体dna的定点突变方法。
背景技术:
基因的定点突变技术是二十世纪生物化学和分子生物学领域里最重要的发现之一,它为人类了解生物体基因的功能,蛋白质及其基序的结构域和功能,由基因突变导致的遗传疾病发生的致病机制提供了强有力的研究手段。基因的定点突变技术可通过多种实验手段完成;其中,聚合酶链式反应(pcr)指导的定点突变技术具有简单,快速,方便等特点,在生物学,农学和医学等领域中应用广泛。pcr介导的定点突变技术目前一般只适合于大小为3kb至10kb载体或质粒中的基因突变,而且,随着载体大小增加,突变的效率明显降低。对于载体大小在10kb以上的基因突变,不仅费时费力,消耗大量资金,而且突变效率极低。由于生物体基因组中包含大量编码蛋白的超大基因,这些基因被克隆在载体中往往超过了10kb,有的甚至将近20kb。利用现有的pcr定点突变技术突变如此大的dna分子时经常感到束手无策。因此,有必要发明一种突变dna超大分子的pcr定点突变技术。
现有技术中,利用包含突变位点的双链dna小分子(或称完全互补引物)和pcr反应,经dpni酶切和感受态细菌转化等实验流程完成dna定点突变的技术,该技术适合分子量较小的dna定点突变,具有简单,快速等特点,stratagene公司生产的定点突变试剂盒就是基于此方法。
但是由于引物自身能够配对结合,pcr产物含量极低,突变效率极低,即便是小分子的dna载体,通常一次实验不能完成,需要反复尝试才可成功;对pcr条件要求高,不方便进行多点突变,插入突变和缺失突变或者此对类突变效率极低。不能用于大载体或大dna分子的定点突变。
另外,利用包含突变位点的一对5'端互补引物和pcr反应,经dpni酶切和感受态细菌转化等实验流程完成dna定点突变的技术。该技术适合分子量较中或小的dna分子定点突变,具有简单,快速以及完成多种突变等特点,此方法是上面现有技术的改进型。
但是由于pcr引物5'端仍有一端分互补序列,导致pcr产物降低,突变效率降低;而在载体较小,反应良好的情况下,反应产物两端会产生多余的重复序列,这时若想获得正确突变体,需要借助另外一种方法即同源重组的方法完成。但是此方法仍然对大载体dna的突变无效。
再者,利用包含突变位点的一对不互补引物完成反向pcr反应(突变位点包含在其中一个引物5'端中),经dpni酶切,pcr产物纯化,产物末端加磷,连接反应和感受态细菌转化等实验完成定点突变的技术。相比较上面两种现有技术,这一方法具有更好的重复性和更高的突变效率,能够完成多位点突变,插入和缺失突变,适合于中小载体的突变。但是此方法较上面现有技术程序更为复杂,dna模板需要dpni酶切,pcr产物的纯化,磷酸化和连接酶连接等步骤。但对大载体dna的突变依然无效。
技术实现要素:
针对上述技术问题,本发明提供一种基于重组酶和超保真dna聚合酶的大载体dna的定点突变方法,解决目前克隆于大载体(10kb以上)上的基因或dna的定点突变的技术难题,或者载体和基因总共大于10kbdna分子的定点突变技术难题。
具体技术方案为:
基于重组酶和超保真dna聚合酶的大载体dna的定点突变方法,包括以下步骤:
(1)定点突变引物设计
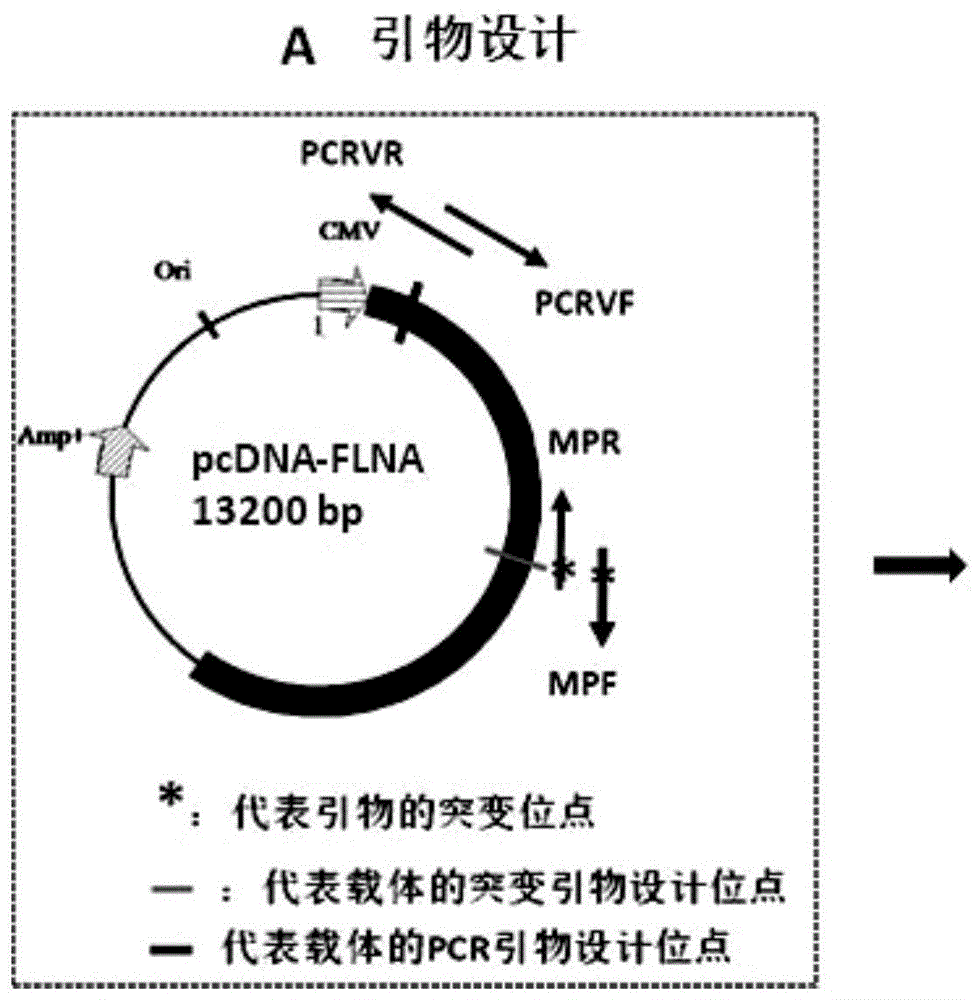
设计两对引物,其中一对为突变引物即mpf和mpr,这对引物的5'端互补序列长度为12-15bp,互补区可以任意引入单点或多点突变,任意长度的缺失突变,插入突变1-20bp;另一对引物是协助pcr的引物,由于这一对引物的位置可以设置在已知序列区内,是可变动的,可以在突变位点的前面或后面设置,称之为pcr可变引物pcrvf和pcrvr(图1a);
(2)聚合酶链式反应设置和产物纯化(i)以0.5-1ngdna为模板,用phantamax超保真dna聚合酶分别设立两个不同的pcr反应;
(ii)pcr热循环;
(iii)利用axygen胶回收试剂盒按照试剂盒说明书回收纯化以上两种pcr产物,利用琼脂糖凝胶电泳对回收产物进行鉴定;
(3)重组酶连接反应
将上述步骤(2)中子步骤(iii)中纯化的dna按照等摩尔数混合,利用重组酶exnaseii完成连接反应;
(4)转化,质粒提取和测序分析。
进一步的,步骤(2)中子步骤(i)两个不同的pcr反应的体系如下:
pcr反应体系i包括:dna模板0.5ng或稀释的dna1μl,引物mpf和引物pcrvr各0.5μl,含phantamax超保真dna聚合酶反应混合物(2x)20μl,双蒸水18.5μl,共40μl;
pcr反应体系ii包括:dna模板0.5ng或稀释的dna1μl,引物pcrvf和引物mpr各0.5μl,含phantamax超保真dna聚合酶反应混合物(2x)20μl,双蒸水18.5μl,共40μl。
其中,步骤(2)中的子步骤(ii)pcr热循环为:(1)95℃3分钟;(2)95℃1分钟,55℃15秒,72℃1-10分钟,取决于pcrdna产物长度,1kb/30秒,35个循环;(3)72℃5分钟。
优选的,步骤(3)所述的连接反应,为20μl反应体系:pcr产物ixμl,pcr产物iiyμl,5x连接酶缓冲液,exnaseii1μl,加ddh2o至20μl,25℃下连接15-20min。
步骤(4)转化,质粒提取和测序分析,具体步骤为:
(i)转化:取细菌感受态细胞30μl置于冰上,加入步骤(3)中的连接产物3μl,冰浴45min;冰浴完成后于42℃水浴锅中热激60sec,置冰上冰浴2min后加入200μlsoc溶液,转至37℃摇床上振荡1h;将转化连接产物的感受态细胞均匀涂在含抗生素的lb固体培养基上并置于37℃恒温箱培养14-16h;
(ii)质粒提取:用接菌环从lb培养皿中挑取单菌落接种于3mllb液体培养基中,转至37℃摇床振荡培养过夜或16h;菌液转入离心管中和在12000rpm速度下离心,收集菌体;利用axygen质粒提取试剂盒按照试剂盒说明书提取质粒;取3μl质粒完成琼脂糖凝胶电泳分析;
(iii)测序分析:取1μg质粒,送测序公司完成dna测序,分析定突变位置的序列是否突变;若每个突变反应完成多个质粒的提取和测序分析,则可分析出此方法完成的每个突变反应的突变效率。
本发明方法具有的优点包括:(1)是一种新颖的针对传统方法不能对付的克隆于大载体/质粒上的基因或者大基因(10kb-20kb)的定点突变方法。(2)不涉及搭桥pcr(overlappingpcr)。(3)不涉及传统基因克隆,而是,pcr产物纯化后,采用重组酶方法连接,一般只需要15-20分钟即可转化,时间短,方法相对简单。(3)针对的是载体dna,只需要两次普通pcr,而搭桥突变方法则针对要克隆的基因,至少完成两次普通pcr,一次搭桥pcr。(4)引物的灵活性很高,采用两对部分互补引物完成,而且,在有需要情况下两对引物都可引入突变位点可加快突变的效率,或者设计一对位置固定的突变引物,另一对引物可以随机变动位置,这样在pcr遇到困难时,可随机改变pcr可变引物的位置,这是其它已发表的方法不具备的特点。(5)利用phantamax超保真酶能够扩增近20kb长度的dna,这也是成功的关键之一,以前的突变方法也没有使用过。
本发明提供的基于重组酶和超保真dna聚合酶的大载体dna的定点突变方法,具有的有益效果:
(1)由于突变引物是分开用于pcr,因此,可在其互补区域取任意置换碱基(一次至少达到15个),进行任意长度的缺失突变,长片段拆入突变,这是其它方法无法达到的。
(2)pcr可变引物,如有需要可随时转换成突变引物,这样可显著提高突变的效率,特别是对于相距较远的多位点突变更有优势,其它方法无法达到此目的。
(3)本发明dna模板用量少,1ng甚至更少便足够;
(4)由于模板始终大于pcr产物不需要dpni酶切便可纯化。
(5)本发明可以用于大载体dna定点突变而且表现出很高突变效率。也适用于中小载体的dna定点突变。
附图说明
图1a为本发明方法的第一步骤示意图;
图1b为本发明方法的第二步骤示意图;
图1c为本发明方法的第三步骤示意图;
图2a为第传统pcr介导的定点突变方法实验流程示意图;
图2b为第二种传统pcr介导的定点突变方法实验流程示意图;
图2c为第三种传统pcr介导的定点突变方法实验流程示意图;
图3a不同定点突变方法所获得的pcr产物琼脂糖凝胶电泳结果;
图3b为不同定点突变方法获得的产物经转化后菌落形成结果;
图3c不同突变方法的阳性克隆的比较。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。通常在此处附图中描述和示出的本发明实施例的组件可以以各种不同的配置来布置和设计。因此,以下对在附图中提供的本发明的实施例的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
基于重组酶和超保真dna聚合酶的大载体dna的定点突变方法,包括以下步骤:
(1)定点突变引物设计
按照图1a所示设计两对引物,其中一对为突变引物(mpf和mpr),这对引物的5'端互补序列长度为12-15bp,互补区可以任意引入单点或多点突变,任意长度的缺失突变,插入突变(1-20bp)。另一对引物是协助pcr的引物,由于这一对引物的位置可以设置在已知序列区内,是可变动的,可以在突变位点的前面或后面设置,称之为pcr可变引物pcrvf和pcrvr。
(2)聚合酶链式反应(pcr)设置和产物纯化
(i)以0.5-1ngdna为模板,利用vazyme公司生产的phantamax超保真dna聚合酶(此酶可以扩增10-20kbdna片段)按照图1b所示分别设立两个不同的pcr反应。反应体系如下:
pcr反应体系i包括:dna模板0.5ng或稀释的dna1μl,引物mpf和引物pcrvr各0.5μl,含phantamax超保真dna聚合酶反应混合物(2x)20μl,双蒸水18.5μl,共40μl。
pcr反应体系ii包括:dna模板0.5ng或稀释的dna1μl,引物pcrvf和引物mpr各0.5μl,含phantamax超保真dna聚合酶反应混合物(2x)20μl,双蒸水18.5μl,共40μl。
(ii)pcr热循环为:(1)95℃3分钟,(2)95℃1分钟,55℃15秒,72℃1-10分钟(取决于pcrdna产物长度,1kb/30秒),35个循环。(3)72℃5分钟。
(iii)利用axygen胶回收试剂盒按照试剂盒说明书回收纯化以上两种pcr产物,利用琼脂糖凝胶电泳对回收产物进行鉴定。
(3)重组酶连接反应
将上述(2)中的(iii)步中纯化的dna按照等摩尔数混合,利用vazyme公司生产的重组酶exnaseii完成连接反应,如图1c:20μl反应体系如下:pcr产物ixμl,pcr产物iiyμl,5x连接酶缓冲液,exnaseii1μl,加ddh2o至20μl。
(4)转化,质粒提取和测序分析
转化,质粒提取和测序分析按照普通分子生物学方法完成,具体如下:
(i)转化:取细菌感受态细胞30μl置于冰上,加入第(3)步中的连接产物3μl,冰浴45min;冰浴完成后于42℃水浴锅中热激60sec,置冰上冰浴2min后加入200μlsoc溶液,转至37℃摇床上振荡1h。将转化连接产物的感受态细胞均匀涂在含抗生素的lb固体培养基上并置于37℃恒温箱培养过夜(14-16h)。
(ii)质粒提取:用接菌环从lb培养皿中挑取单菌落接种于3mllb液体培养基中,转至37℃摇床振荡培养过夜或16h。菌液转入离心管中和在12000rpm速度下离心,收集菌体。利用axygen质粒提取试剂盒按照试剂盒说明书提取质粒。取3μl质粒完成琼脂糖凝胶电泳分析。
(iii)测序分析:取1微克质粒,送测序公司完成dna测序,分析定突变位置的序列是否突变。若每个突变反应完成多个质粒的提取和测序分析,则可分析出此方法完成的每个突变反应的突变效率。
为了说明本发明的突变效果以及与其他传统定点突变方法的比较分析及其优势
本发明提供应用例:克隆于pcdna载体中的细丝蛋白a基因(flna)(质粒dna共13.2kb)定点突变效果分析
(一)材料:突变引物:flnamf,ctgctttgccgcatccaaagtcaagtg
;flnamr,ggatgcggcaaagcagggaaccacg
pcr引物:pcdnapcrf,actctcgggcgggccagagcgcagcag;pcdnapcrr,cccgcccgagagtgggagctactcat.
dna模板:pcdna-flna质粒;
pcr反应混合物购自vazyme生产的2xphantamastermix(货号.p515-01)。重组连接酶购自vazyme生产的克隆重组酶(货号.c112-01)。感受态细胞购自擎科生物公司(武汉),测序分析由擎科生物公司完成。
(二)方法:
按照本发明方法的步骤,利用1ngpcdna-flna为模板,引物flnamf与pcdnapcrr完成pcr反应i,利用引物pcdnapcrf与flnamr完成pcr反应ii,将pcr产物进行纯化,利用重组连接酶exnaseii将两种pcr产物进行连接,转化,质粒提取和测序分析,如图1a到图1c所示。
传统定点突变的方法按照图2a、图2b和图2c的步骤方法进行。
(三)突变效果分析
图3a不同定点突变方法所获得的pcr产物琼脂糖凝胶电泳结果。图3b为不同定点突变方法获得的产物经转化后菌落形成结果。图3c不同突变方法的阳性克隆的比较。
图中a代表传统定点突变方法a(图2a),b代表传统定点突变方法b(图2b),c代表传统定点突变方法c(图2c),p1和p2分别代表本发明分获得的pcr产物1和2.p1+p2代表本发明方法。
(i)本发明方法很容易获得pcr产物
以pcdna-flna为模板,利用本发明的方法和其它传统方法分别完成聚合聚合酶链式反应,分别取取5微升样品完成琼脂糖凝胶电泳并在紫外灯下成像。如图3a所示,本发明的方法通过pcr能够扩增出产物,而三种传统定点突变方法,不管pcr引物是否互补,均没有获得pcr产物。
(ii)本发明方法制备的连接产物经转化后能够产生抗性菌落
在通常情况下,传统定点突变方法(图2a和图2b)琼脂糖凝胶电泳后无法观察到pcr产物,但不能由此否定其定点突变不成功,因为微量环状质粒可能通过转化而呈现。因此,将上述第(i)步中的产物分别按照各自的方法处理后转化dh5α感受态细胞,第二天观察其产物的转化效率,图3b所示,采用本发明的方法完成的转化获得较多数量的菌落,说明本发明制备的连接产物可能获得有抗性的质粒;而其它定点突变方法获得的产物经转化后没有获得菌落,说明以前报道的定点突变方法对于大载体dna的定点突变无效。
(iii)本发明方法具有很高突变效率
虽然在第(ii)步中,观察到本发明方法获得的连接产物经转化后获得了不少菌落,但其突变效率并不清楚。因此,将上述菌落接种于lb培养基,37℃摇床振荡过夜,第二天收集菌体,提取质粒,将质粒完成测序分析。结果如图3c所示,本发明的方法突变效率达到100%。其它定点突变方法,如第(ii)步所描述的,对于大载体dna的突变完全没有效率。
以上所述仅为本申请的优选实施例而已,并不用于限制本申请,对于本领域的技术人员来说,本申请可以有各种更改和变化。凡在本申请的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本申请的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!