用于增强基因组覆盖和保持空间邻近的邻接性的方法和组合物与流程
用于增强基因组覆盖和保持空间邻近的邻接性的方法和组合物
1.相关专利申请
2.本技术要求于2019年5月20日提交的名称为“用于增强基因组覆盖的方法和组合物(methods and compositions for enhanced genome coverage)”、发明人名字为anthony schmitt、derek reid、stephen mac、xiang zhou和siddarth selvaraj且指定代理人案卷号为amg-1004-pv的美国临时专利申请号62/850,449的权益。本技术涉及于2019年11月19日提交的名称为“用于制备保持空间邻近的邻接性信息的核酸的方法(methods for preparing nucleic acids that preserve spatial-proximal contiguity information)”、发明人名字为anthony schmitt、catherine tan、derek reid、chris de la torre和siddarth selvaraj且指定代理人案卷号为amg-1003-ut的美国专利申请号16/689,002。本技术还涉及于2020年5月15日提交的名称为“在核酸模板中保持空间邻近的邻接性和分子邻接性(preserving spatial-proximal contiguity and molecular contiguity in nucleic acid templates)”、发明人名字为siddarth selvaraj、anthony schmitt和bret reid且指定代理人案卷号为amg-1002-us的美国专利申请号16/764,787。本技术还涉及于2017年12月21日提交的名称为“混合物样品的精确分子解卷积(accurate molecular deconvolution of mixture samples)”、发明人名字为siddarth selvaraj、nathaniel heintzman和christian edgar laing且指定代理人案卷号为amg-1001-us的美国专利申请号15/738,871。上述专利申请的全部内容以引用方式并入本文,包括所有文本、表和附图。
3.政府支持声明
4.本发明是在政府支持下在美国国家卫生研究院(the national institutes of health)授予的合同号1r44hg009584-01和2r44hg008118-04a1拨款下作出的。政府拥有本发明中的某些权利。
技术领域
5.本技术部分涉及对核酸进行测序。
背景技术:
6.下一代测序(ngs)已成为用于确定用于众多研究和临床应用的核酸序列的主要方法集。典型的ngs工作流程如下:将通常被组织为一个或多个染色体的天然基因组dna从导致其片段化的核酸源中分离出来,以产生核酸模板,随后通过测序仪读取所述核酸模板以生成序列数据。
技术实现要素:
7.本技术涉及通过以下方式制备dna分子的方法:保持空间邻近的邻接性信息并提供等同于全基因组测序的覆盖的完全基因组覆盖。
8.在某些方面,提供了一种用于从样品制备dna分子的方法,其包括:
9.(a)使包括基因组或其部分的样品的交联dna分子与一组限制性核酸内切酶接触;从而生成交联dna分子的空间邻近的消化末端;(b)使交联dna分子的所述空间邻近的消化末端与连接酶接触,从而生成包括连接接点的交联的邻位连接dna分子;(c)使包括连接接点的所述交联的邻位连接dna分子与逆转交联的试剂接触,从而生成包括连接接点的邻位连接dna分子;以及(d)将所述邻位连接dna分子片段化,以生成邻位连接dna分子的片段,所述邻位连接dna分子的片段包括跨越所述连接接点的片段,其中跨越所述连接接点且长度可为用于短程测序的模板的片段包括基本上全基因组或其部分的序列。
10.在某些方面,还提供了一种用于从样品制备dna分子的方法,其包括:(a)使包括基因组或其部分的样品的交联dna分子与第一限制性核酸内切酶接触,从而生成交联dna分子的第一空间邻近的消化末端;(b)使交联dna分子的所述第一空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点的第一交联邻位连接dna分子;(c)使包括第一连接接点的所述第一交联邻位连接dna分子与第二限制性核酸内切酶接触,从而生成交联dna分子的第二空间邻近的消化末端;(d)使交联dna分子的所述第二空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点和第二连接接点的第二交联邻位连接dna分子;(d)使交联dna分子的所述第二空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点和第二连接接点的第二交联邻位连接dna分子;(e)使包括第一连接接点和第二连接接点的所述第二交联邻位连接dna分子与第三限制性核酸内切酶接触,从而生成交联dna分子的第三空间邻近的消化末端;(f)使交联dna分子的所述第三空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点、第二连接接点和第三连接接点的第三交联邻位连接dna分子;(g)使包括第一连接接点、第二连接接点和第三连接接点的所述第三交联邻位连接dna分子与第四限制性核酸内切酶接触,从而生成交联dna分子的第四空间邻近的消化末端;(h)使交联dna分子的所述第四空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点、第二连接接点、第三连接接点和第四连接接点的第四交联邻位连接dna分子;(i)使包括第一连接接点、第二连接接点、第三连接接点和第四连接接点的所述第四交联邻位连接dna分子与逆转交联的试剂接触,从而生成包括第一连接接点、第二连接接点、第三连接接点和第四连接接点的邻位连接dna分子;以及(j)将所述邻位连接dna分子片段化,以生成邻位连接dna分子的片段,所述邻位连接dna分子的片段包括跨越所述第一连接接点、所述第二连接接点、所述第三连接接点和所述第四连接接点的片段,其中跨越所述第一连接接点、所述第二连接接点、所述第三连接接点和所述第四连接接点且长度可为用于短程测序的模板的片段包括基本上全基因组或其部分的序列。
11.在某些方面,还提供了一种用于从样品制备dna分子的方法,其包括:(a)使包括基因组或其部分的样品的交联dna分子与一组四种限制性核酸内切酶接触;从而生成交联dna分子的空间邻近的消化末端;(b)使交联dna分子的所述空间邻近的消化末端与一种或多种试剂接触,所述一种或多种试剂将附接至核苷酸的生物素掺入到空间邻近的消化末端中,从而生成包括有标记的空间邻近的消化末端的交联dna分子;(c)使包括标记的空间邻近的消化末端的所述交联dna分子与连接酶接触,从而生成包括标记的连接接点的交联邻位连接dna分子;(d)使包括标记的连接接点的交联邻位连接dna分子与逆转交联的试剂接触,从而生成包括标记的连接接点的邻位连接dna分子;(e)将包括标记的连接接点的所述邻位连
接dna分子片段化,以生成邻位连接dna分子的片段,所述邻位连接dna分子的片段包括跨越所述标记的连接接点的片段,其中跨越所述连接接点且长度可为用于短程测序的模板的片段包括基本上全基因组或其部分的序列;以及(f)通过使用包括链霉抗生物素蛋白的亲和纯化分子对标记的连接接点进行亲和纯化,富集跨越所述标记的连接接点的dna片段。
12.在某些方面,还提供了一种用于从样品制备dna分子的方法,其包括:(a)使来自样品的具有稳定空间相互作用的空间邻近的dna分子与两种或更多种限制性核酸内切酶接触,从而消化所述dna分子,并生成dna分子的空间邻近的消化末端;以及(b)使dna分子的所述空间邻近的消化末端与连接酶接触,从而生成包括连接接点的邻位连接dna分子,其中所述连接接点是未标记的。
13.在某些方面,还提供了一种用于从样品制备dna分子的方法,其包括:(a)使来自样品的细胞/细胞核内具有稳定空间相互作用的空间邻近的dna分子与两种或更多种限制性核酸内切酶接触,从而消化所述dna分子,并生成dna分子的空间邻近的消化末端;以及(b)使dna分子的所述空间邻近的消化末端与连接酶接触,从而生成包括连接接点的邻位连接dna分子,其中所述连接接点是未标记的,并且所述接触步骤在原位进行。
14.在某些方面,还提供了一种用于从样品制备dna分子的方法,其包括:(a)使来自样品的具有稳定空间相互作用的空间邻近的dna分子与第一限制性核酸内切酶接触,从而消化所述dna分子,并生成dna分子的第一空间邻近的消化末端;(b)使dna分子的所述第一空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点的第一邻位连接dna分子,其中所述连接接点是未标记的;(c)使包括第一连接接点的所述第一邻位连接dna分子与第二限制性核酸内切酶接触,从而消化所述第一邻位连接dna分子,并生成dna分子的第二空间邻近的消化末端;以及(d)使dna分子的所述第二空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点和第二连接接点的第二邻位连接dna分子,其中所述连接接点是未标记的。
15.在某些方面,还提供了一种方法,其中(e)使包括第一连接接点和第二连接接点的第二邻位连接dna分子与第三限制性核酸内切酶接触,从而消化所述第二邻位连接dna分子,并生成dna分子的第三空间邻近的消化末端;以及(f)使dna分子的所述第三空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点、第二连接接点和第三连接接点的第三邻位连接dna分子,其中所述连接接点是未标记的。
16.在某些方面,还提供了一种用于从样品制备dna分子的方法,其包括:(a)使在来自样品的细胞/细胞核内具有稳定空间相互作用的空间邻近的dna分子与第一限制性核酸内切酶接触,从而消化所述dna分子,并生成dna分子的第一空间邻近的消化末端;(b)使dna分子的所述第一空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点的第一邻位连接dna分子,其中所述连接接点是未标记的,并且所述接触步骤在原位进行;(c)使包括第一连接接点的所述第一邻位连接dna分子与第二限制性核酸内切酶接触,从而消化所述第一邻位连接dna分子,并生成dna分子的第二空间邻近的消化末端;以及(d)使dna分子的所述第二空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点和第二连接接点的第二邻位连接dna分子,其中所述连接接点是未标记的,并且所述接触步骤在原位进行。
17.在某些方面,还提供了一种方法,其中(e)使包括第一连接接点和第二连接接点的第二邻位连接dna分子与第三限制性核酸内切酶接触,从而消化所述第二邻位连接dna分
子,并生成dna分子的第三空间邻近的消化末端;以及(f)使dna分子的所述第三空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点、第二连接接点和第三连接接点的第三邻位连接dna分子,其中所述连接接点是未标记的,并且所述接触步骤在原位进行。
18.在某些方面,还提供了利用上述优化的3c方案的方法,其应用受益于含有连接接点的读段对的增加的覆盖均匀性,所述应用诸如对基因组、宏基因组组装体中的重叠群进行聚类、排序和定向以及单倍型定相。
19.在某些方面,还提供了利用上述优化的3c方案用于以下应用的方法,其应用依赖于1d基因组覆盖均匀性,诸如snv发现、断点检测、碱基校正(polish)基因组组装和1d“峰值识别”,诸如在chip-seq中。
20.在某些方面,还提供了利用上述优化的3c方案用于以下应用的方法,其应用受益于增加的连接事件,所述增加的连接事件保持了空间邻近的邻接性信息,所述应用诸如检测成对的3d基因组相互作用和3d构象分析。
21.在某些方面,还提供了利用本文所述方法制备的文库。
22.在某些方面,还提供了试剂盒,其包括用于进行本文所述方法的试剂。
23.在某些方面,还提供了获得从邻位连接组织切片3c或hic得到的序列信息的空间定位的方法。
24.在以下描述、实施例、权利要求和附图中进一步描述了某些实施方案。
附图说明
25.附图示出了本技术的某些实施方案,且不具有限制性。为了清楚和易于说明,附图不是按比例绘制的,并且在一些情况下,多个方面可被夸大或放大显示,以便于理解特定实施方案。
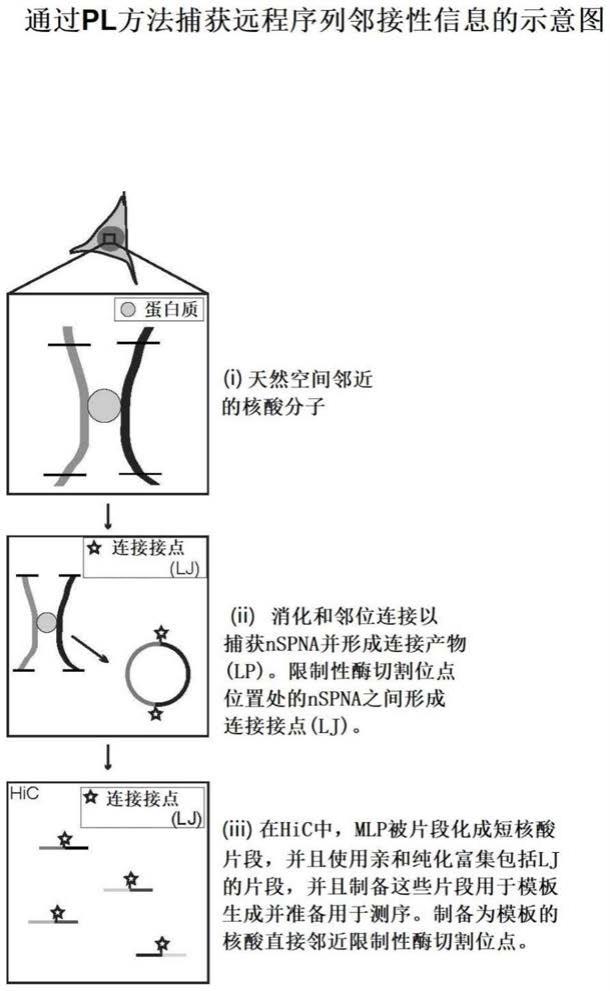
26.图1显示通过pl(邻位连接)方法捕获空间邻近的邻接性信息。
27.图2a和图2b显示超高re切割位点密度使得能够进行均匀的基因组覆盖。
28.图3a和图3b显示最佳限制性酶的选择。
29.图4显示与四位个体中的鸟枪法(shotgun)全基因组测序(wgs)相比等同的snv发现性能。
30.图5a和图5b示出了更精确的基因组重排断点检测。
31.图6a至图6d示出了更全面的重叠群(contig)聚类和更精确的重叠群排序。
32.图7示出了更精确的重叠群定向。
33.图8a和图8b示出了更高分辨率的3d基因组构象分析。
34.图9示出了高度灵敏的蛋白质因子定位和3d构象分析。
35.图10示出了高度灵敏且同时的变体发现和单倍型定相分析。
36.图11示出了通过实现为同时消化的多酶3c而改善的核酸模板中空间邻近的邻接性的保持。
37.图12示出了通过实现为顺序消化的多酶3c而改善的核酸模板中空间邻近的邻接性的保持。
38.图13示出了通过3c文库中大片段的大小选择而改善的核酸模板中空间邻近的邻接性的保持。
39.图14a和图14b示出了hicoverage使得能够进行跨整个植物和动物物种范围的接近完全的基因组覆盖。图14a涉及脊椎动物基因组。图14b涉及昆虫基因组、植物基因组和寄生虫基因组。
40.图15示出了hicoverage使得能够进行均匀的基因组覆盖。
41.图16a和图16b示出了通过实现为顺序轮次的消化和连接的多酶3c而改善的含有连接接点的核酸模板的空间邻近的邻接性的保持和基因组覆盖。图16a示出了消化和连接的产物的大小。图16b示出了远程顺式读出(read-out)的百分比。
具体实施方式
42.本文提供用于制备测序模板的方法和组合物,所述方法和组合物提供均匀的基因组覆盖并保持空间邻近的邻接性信息。
43.邻位连接
44.pl方法(参见图1)始于(i)交联的核酸源(例如细胞核、细胞、组织、ffpe样品)内天然空间邻近的核酸(nspna)),之后进行(ii)溶解和解压缩(decompacted)的样品的染色质的消化(例如通过re,参见黑色刻度标记(tick mark))以及空间邻近的消化末端的连接,以生成连接产物(lp),由此连接接点显现在来自每个连接的nspna的相应re切割位点位置处并保持空间邻近的邻接性信息。广义地说,将pl方法分类为基于3c的和基于hic的,但pl有多种特定变化形式。
45.在3c中,多个lp被片段化,制备成短核酸模板并且准备用于测序。在3c中,核酸模板包括邻近re切割位点和远离re切割位点的核酸(dekker等人,science 295,1306-1311(2002))。
46.在hic中,对消化的核酸末端进行标记(例如生物素化),然后连接以产生在连接接点(lj)处带有亲和纯化标记物的经标记的连接产物(mlp,mlp是lp的显现形式)。在将多个mlp片段化之后,使用亲和纯化来富集包括lj的mlp片段,并且将这些片段制备为核酸模板并准备用于测序,即将来自至少含有lj的mlp的片段化的核酸富集并制备为模板,并且在hic中测序,以耗尽umlp(通常不显现lj的未连接mlp)。由于对lj的这一富集,因此核酸模板仅包括邻近re切割位点的核酸(参见lieberman-aiden等人,us2017/0362649、lieberman-aiden等人,science 326,289-293(2009)、dekker等人(美国专利号9434985))。
47.在一些实施方案中,邻位连接方法通常包括以下步骤:(1)用限制性酶消化(或片段化)溶解和解压缩的样品的染色质;(2)使消化或片段化的末端变平或省略变平过程;以及(3)连接空间邻近的末端,从而保持空间邻近的邻接性信息。在保持了空间邻近的邻接性信息后,进一步的步骤可以包括:使用大小选择来纯化和富集代表连接接点片段的连接片段,由富集的片段制备文库以及对文库进行测序。
48.在一些实施方案中,邻位连接的核酸分子是在原位生成的。如本文所用的术语“原位”是指在细胞核内(参见美国专利申请us2017/0362649)。
49.在一些实施方案中,在除了3c或hic之外的染色质构象测定中分析邻位连接dna分子。在一些实施方案中,染色质构象测定是capture-c(hughes等人,nature genetics,46(2),第205页(2014))、4c(simonis等人,nature genetics 38,1348-1354(2006),de laat等人(u.s.8642295))、5c(dostie等人,genome research 16,1299-1309(2006),dekker等
人(us9273309))、capture-hic(等人,nature communications,6,第6178页(2015))、hichip(mumbach等人,nature methods,13(11),第919-922页(2016))、plac-seq(fang等人,cell research,26(12),第1345-1348页(2016))、栓系染色体捕获(tethered chromosome capture)(tcc)(kalhor等人,nature biotechnology 30,90-98(2012),chen等人(us20110287947))、hiculfite(stamenova等人,biorxiv,第481283页(2018))、methy-hic(li等人,nature methods,16(10),第991-993页(2019))、hichirp(mumbach等人,nature methods,16(6),第489-492页(2019))或其组合。
50.不管具体的pl方法如何,所有的pl方法都以连接产物的形式捕获空间邻近的邻接性信息,由此在两个天然的空间邻近的核酸之间形成连接接点。在形成lp后,则使用下一代测序来检测空间邻近的邻接性信息,由此对一个或多个连接接点(来自整个lp或lp的片段)进行测序(如本文所述)。利用这些序列信息,可以得知来自给定连接产物(或连接接点)的核酸分子是天然的空间邻近的核酸。
51.在某些实施方案中,其中测定是全基因组的(即,针对全基因组)。
52.在一些实施方案中,测定是3c、hic、栓系染色体捕获(tcc)、hiculfite、methy-hic或其组合。
53.在某些实施方案中,测定针对基因组中的一个或多个靶区域。在一些实施方案中,测定是capture-c、4c、5c、capture-hic、hichip、plac-seq、hichlrp或其组合。在一些实施方案中,靶标是单核苷酸变异、插入、缺失、拷贝数变异、基因组重排或用于定相的靶标。
54.在一些实施方案中,样品包括癌症基因组,并且靶区域与癌症的表型相关。在一些实施方案中,与癌症相关的靶标是结构变异,诸如基因组重排或拷贝数变异。在某些实施方案中,靶标是癌基因或一组癌基因。
55.超高切割位点密度
56.图2a和图2b显示通过使邻近re切割位点且因此将在hic核酸测序模板中表示的核酸的量最大化(超高切割位点密度“hicoverage”)来使基因组覆盖最大化。图2a是显示基于人基因组(hg19)的re基序、理论re消化频率和计算机中的(in silico)平均消化频率的表。在本文所述的方法中,在hic期间,使用多种re 4-切割酶(4-cutter)的混合剂(cocktail)同时消化基因组。这相较于标准hic方案将re切割位置密度增加了一个对数级,并且这样使基因组覆盖和均匀性最大化到可与鸟枪法wgs相当的水平(参见图2b),并且使得能够进行受益于或需要均匀的全基因组覆盖的数据应用(还参见实施例3以及图14a和图14b,跨整个植物和动物物种范围的完全的基因组覆盖;以及实施例4和图15,覆盖均匀性)。最大化的基因组覆盖和均匀性表示在跨越连接接点的邻位连接dna分子的片段中。连接接点在基因组中的分布是所述方法的超高切割位点密度的结果。跨越连接接点的邻位连接dna分子的片段包括基本上全基因组或其一部分的序列。在一些实施方案中,跨越连接接点且长度可为用于短程测序的模板的片段包括基本上全基因组或其部分的序列。在某些实施方案中,跨越连接接点的片段包括多达750个碱基对的片段。
57.限制性核酸内切酶
58.在一些实施方案中,所述方法中所用的限制性核酸内切酶各自具有约1/256的理论消化频率,并且当组合四种限制性核酸内切酶时,具有约1/64的理论消化频率。然而,在理论消化频率、预测的计算机中的频率和染色质消化后观察到的片段大小之间存在差异。
理论消化频率和计算机中的频率是给定限制性核酸内切酶将如何消化染色质且特别是交联染色质的差预测指标。
59.在一些实施方案中,使样品的交联dna分子与一组限制性核酸内切酶接触,使得每种限制性核酸内切酶在大约相同的时段期间发挥消化交联dna分子的作用。在一些实施方案中,一组限制性核酸内切酶各自在常用缓冲液中具有高活性水平(即,大约100%的最佳切割效率)。常用缓冲液的示例是cutsmart
tm
(new england biolabs,beverly,ma)。
60.在一些实施方案中,限制性核酸内切酶可以产生具有5’突出端、3’突出端或无突出端(即平末端)的dna分子。
61.在一些实施方案中,一组限制性核酸内切酶可以是至少三种限制性核酸内切酶。在某些实施方案中,一组限制性核酸内切酶由四种限制性核酸内切酶组成。在一些实施方案中,样品包括不同于细菌基因组的基因组,并且选择一组限制性核酸内切酶来消化该基因组。在某些实施方案中,四种限制性核酸内切酶是:mboi、hinfi、msei和ddei。在一些实施方案中,如宏基因组样品中,样品包括一个或多个细菌基因组,并且选择一组限制性核酸内切酶来消化该一个或多个细菌基因组。在某些实施方案中,四种限制性核酸内切酶是:hpych4iv、hinfi、hinp1i和msei。
62.在一些实施方案中,限制性核酸内切酶可以按顺序添加到样品中,并且不同时消化样品中的交联dna分子。在一些实施方案中,限制性核酸内切酶生成具有相同类型末端的dna分子。在一些实施方案中,限制性核酸内切酶中的两种或更多种生成具有不同类型末端(例如,5’突出端、3’突出端、无突出端或平端)的dna分子。在一些实施方案中,限制性核酸内切酶中的一种或多种需要用于高活性水平的特定缓冲液,该特定缓冲液不同于另一种限制性核酸内切酶的高活性水平所需的缓冲液。当限制性核酸内切酶单独接触样品中的交联dna分子时,如果需要,可以向每种限制性核酸内切酶提供其自身独特的缓冲液。在某些实施方案中,按顺序添加到样品中的限制性核酸内切酶可以生成消化末端,所述消化末端可以掺入标记核苷酸,所述标记核苷酸不同于被掺入到由不同限制性内切酶生成的消化末端中的标记核苷酸。这与同时消化样品dna分子的限制性核酸内切酶的使用不同,所述限制性核酸内切酶限于在各种消化末端掺入常用的标记核苷酸。
63.测序
64.核酸模板(或简称为“模板”)是指通过测序仪读取的一个或多个核酸分子。生成核酸模板的方法通常涉及核酸片段化成推荐用于特定测序仪器的分子长度。例如,现行illumina短读测序可适应高达大约750bp的核酸长度(序列模板分子)。虽然可以利用较小的序列模板分子,但由于增加离切割位点较远的序列覆盖应使基因组覆盖最大化,因此通常使用高达大约750bp的模板分子。模板包括跨越连接接点的片段,并且可以获得连接接点两侧的序列信息。然而,由于dna剪切或片段化是随机的,因此连接接点可以发生在沿着模板分子的任何点处。在一些情况下,它可能非常靠近分子的末端,使得在接点的一侧只有约20bp,且在接点的另一侧有数百bp。接点还可以发生在模板的中间,使得在连接接点的每一侧有数/几百个碱基对。
65.读段(read)长度可以是任何长度,包括但不限于2
×
150bp、2
×
100bp、2
×
75bp或2
×
50bp。
66.在一些实施方案中,为了使获得的跨越连接接点的序列信息量最大,使片段化的
邻位连接分子富集包括连接接点的片段化的邻位连接dna分子,并且使用包括连接接点的片段化的邻位连接dna分子来制备用于dna测序的模板分子库。在某些实施方案中,用亲和纯化标记物标记连接接点。在一些实施方案中,亲和纯化标记物是与核苷酸缀合的生物素。在一些实施方案中,用聚合酶(诸如克列诺大片段(klenow large fragment)),使用单一标记的核苷酸(生物素标记的核苷酸)和其它未标记的核苷酸来补平具有5’突出端的空间邻近的消化末端。在一些实施方案中,可使用酶(诸如t4 dna聚合酶)和生物素标记的所有四种核苷酸对具有3’突出端的空间邻近的消化末端进行末端标记。在某些实施方案中,通过用亲和纯化分子对亲和纯化标记物的亲和纯化进行富集。在一些实施方案中,用亲和纯化分子对亲和纯化标记物的亲和纯化用于hic、capture-hic、hichip、plac-seq、hiculfite或methyl-hic。在一些实施方案中,亲和纯化分子是链霉抗生物素蛋白。在某些实施方案中,链霉抗生物素蛋白包括包被在磁珠上的链霉抗生物素蛋白。
67.在某些实施方案中,包括连接接点的片段化的邻位连接dna分子的富集不利用掺入连接接点中的标记。在一些实施方案中,具有5’突出端或3’突出端的分子末端可以在不加标记的情况下变平,并且可以通过大小选择来富集。在连接步骤之后,代表具有连接接点的邻位连接分子的任何dna分子将大于未连接但被消化的片段。在一些实施方案中,将通过大小选择而富集的包括连接接点的邻位连接dna分子用于3c-seq、4c-seq 5c或capture-c中。
68.在一些实施方案中,模板分子文库提供基因组或其部分的均匀的全基因组覆盖。在一些实施方案中,对模板分子文库进行测序以生成包括序列信息的序列读段。在某些实施方案中,测序是短读测序。
69.在一些实施方案中,序列信息用于基因组的分析中。在一些实施方案中,序列信息用于分析基因组的一部分,例如在靶向测定中。在分析基因组和分析基因组的一部分两者中,覆盖的均匀性和范围是相同的。
70.在一些实施方案中,序列信息用于基因组重排分析、断点鉴别、重叠群的聚类和排序、确定重叠群定向、重叠群的聚类、排序和定向、成对3d基因组相互作用的检测(诸如3d基因组相互作用在启动子、增强子、基因调控元件、gwas基因座、染色质环和拓扑结构域锚、重复元件、多梳区、基因体、外显子或整合的病毒序列之间)、蛋白质因子定位分析和3d构象、包括plac-seq或hichip的蛋白质因子定位分析和3d构象分析、单倍型定相、基因组组装和3d构象分析、dna甲基化分析、dna甲基化分析和3d基因组相互作用的检测、单核苷酸变体(snv)发现、对远程测序信息进行碱基校正、高灵敏度拷贝数变异(cnv)分析(例如,拷贝数变异(cnv)是扩增,拷贝数变异(cnv)是杂合或纯合缺失)、变体发现、单倍型定相和基因组组装、单倍型定相和基因组组装、基因组组装和3d基因组相互作用的检测或其组合。
71.在某些实施方案中,序列信息用于包括父本基因组的第一样品和包括母本基因组的第二样品中的变体发现和单倍型定相,并且父本基因组和母本基因组的定相变体用于分析从母体的cfdna中获得的胎儿基因组的序列数据。
72.通过本文所述的方法获得的完全基因组覆盖和空间邻近的邻接性信息可用于利用此类序列信息的其它方法或方法的组合中。
73.样品
74.在一些实施方案中,从选自细胞核、细胞、组织、福尔马林(formalin)固定的石蜡
包埋(ffpe)样品、深度福尔马林固定的样品或无细胞dna的样品中获得dna。在某些实施方案中,从单个细胞获得dna。在某些实施方案中,从两个或更多个细胞中获得dna。在一些实施方案中,样品可以包括两个或更多个代表不同物种的基因组,诸如在宏基因组样品中。
75.基因组重排断点检测
76.图5a和图5b显示与先前的hic方法相比,超高re切割位点密度(hicoverage)如何实现更精确的基因组重排分析。在图5a中,当re切割位点密度低时,诸如在先前的hic方法(lieberman-aiden,science,2009;rao,cell,(2014))中,在含有连接接点的核酸模板中显现的远程“链接(link)”(参见弧线)通过捕获跨越基因组断点的信号来告知基因组重排断点的近似位置。虽然这有助于限定断点的近似位置,但没有跨越断点的核酸模板分子,因为断点位于re切割位点远端,因此此类分析的精度受到序列覆盖的限制。在图5b中,超高re切割位点密度(hicoverage)还包括在含有连接接点的核酸模板中显现的远程“链接”(参见弧线),其通过捕获跨越基因组断点的信号来告知基因组重排断点的近似位置,而且增加的re切割位点密度允许跨越基因组重排断点的嵌合核酸模板分子,以使得能够进行断点精度分析。
77.重叠群聚类和排序
78.图6a至图6d显示在超高re切割位点密度(hicoverage)中使基因组覆盖最大化如何独特地使得能够将重叠群更全面(即更完整)地聚类到染色体中,并因此能够在基因组(或宏基因组)组装中进行更准确的重叠群排序。在一种场景中,从头基因组组装工作流程通常涉及以下的组合:进行长读测序技术(例如oxford nanopore,uk)以产生最连续的序列(“重叠群”),之后进行hic。hic数据(来自连接接点的序列信息)的第一种功能是使用在含有连接接点的核酸模板中的重叠群之间的远程“链接”(参见弧线)来告知哪些重叠群来源自基因组组装情况下的同一染色体、或宏基因组组装情况下的同一生物体。因此,认为hic数据使重叠群“聚类(cluster)”。在聚类后,使用重叠群之间的成对远程“链接”的频率,基于以下前提来确定重叠群沿染色体的相对排序:由hic捕获的频繁出现的空间邻近的重叠群由于聚合物的物理性质而也应当是线性邻近的。在图6a中,由现有hic方法产生的低re切割位点密度可导致某些重叠群没有re切割位点,然后在核酸模板或测序数据中未表示。当使用重叠群之间的远程“链接”将重叠群聚类到一个或多个染色体中时,没有re切割位点的重叠群不能被聚类,从而导致不全面或不完全的染色体序列组成。作为这种不完全聚类的副产物,重叠群的排序也将是不正确的。在图6a中,重叠群c和d、以及a和c具有最频繁的重叠群间链接,而a和d具有最少的链接。使用该信息,在图6b中,此类重叠群的顺序可以被推断为acd,而b被排除,因此产生错误的重叠群顺序。在图6c中,通过超高re切割位点密度(hicoverage)的覆盖均匀性使得能够捕获所有重叠群之间的远程重叠群间链接,从而使得能够将重叠群全面和完全地聚类到染色体。在图6d中,由于完整的重叠群聚类,因此所有重叠群均可用于基于重叠群间链接频率分析重叠群顺序,并且可以推导出正确的重叠群顺序(abcd)。
79.重叠群定向
80.图7a至图7d显示在超高re切割位点密度(hicoverage)中使基因组覆盖最大化如何独特地使得能够进行更精确的重叠群定向分析。在先前描述的从头基因组组装场景(参见图6a至图6d)中,在重叠群排序分析之后,hic数据的下一个效用是重叠群定向分析,以确
定相邻重叠群的哪些末端应当接合。这可以通过分析相邻重叠群的末端之间的链接的频率来确定,并且也是基于以下前提:由hic捕获的频繁出现的重叠群间链接由于聚合物的物理性质而也应当是线性邻近的。换句话说,具有最高重叠群间链接频率的两个相邻的重叠群末端应该以这两个末端被接合的方式进行定向。为了说明这一概念,在图7至图7d中显示中央重叠群和两个相邻重叠群之间的重叠群间hic链接信息。重叠群的每一个末端均以字母标记,以将id分配给每个重叠群末端,并且将正确的顺序描绘为具有重叠群间hic链接频率信息的abcdef(参见弧线)。在图7a中,不频繁和不均匀的re切割位点密度导致仅来自中央重叠群的左末端的重叠群间hic链接。重叠群间链接频率在c和e之间,而不是c和b之间最大,告知重叠群末端c和e应该被错误地接合,产生不正确的重叠群定向(abdcef)(参见图7b)。在图7c中,通过超高re切割位点密度(hicoverage)的覆盖均匀性使得能够实现源于中央重叠群以及相邻重叠群的更大重叠群间hic链接,使得来自末端c和d的链接信息现在可以告知重叠群定向分析。顶部圆弧描绘源于d的重叠群间hic链接,并且下部圆弧描绘源于c的重叠群间hic链接。如所描绘的,b和c之间以及d和e之间的重叠群间链接频率最大(各自n=6),从而告知那些末端对应该接合,产生正确的重叠群定向(abcdef)(参见图7d)。
81.3d基因组构象
82.图8a和图8b显示在超高re切割位点密度(hicoverage)中使基因组覆盖最大化如何独特地使得能够进行成对3d基因组相互作用的最高分辨率和最灵敏的检测。在使用hic的3d基因组组织分析中,在分析任两个箱(bin)之间的箱对相互作用频率之前,较低分辨率的hic通常被聚集到固定间隔的“箱”中。由hic提供的最高分辨率分析是“限制性片段”水平的hic分析,借此对单个限制性片段之间的成对相互作用频率进行定量,并因此由re切割位点的频率来进行界定。然而,当进行3d基因组分析时,具有相对低的re频率的re可能出现低分辨率和不精确。在一种场景中(参见图8a),含有启动子的限制性片段似乎频繁地与包含两个基因调控元件(推定增强子)的另一下游限制性片段相互作用。由于在限制性片段内含有两个增强子,因此不清楚哪个增强子将调控基因a。在图8b中,相同总数的相互作用源于含有基因a启动子的限制性片段,然而,它们由于较高的re切割位点密度而现在与更多相邻的限制性片段链接。如所描绘的,最频繁的相互作用是与包含推定2号增强子的限制性片段的相互作用,这有助于将其鉴别为基因a的靶增强子,而不是相邻的推定3号增强子。注意,启动子-增强子相互作用的成对检测仅代表一种类型的3d相互作用分析。其它分析包括但不限于启动子、增强子、其它基因调控元件、gwas基因座、染色质环和拓扑结构域锚、以及其它所关注的基因组元件或序列(例如重复元件、多梳区、基因体、外显子、整合的病毒序列等)之间的成对相互作用。
83.在超高re切割位点密度(hicoverage)中使基因组覆盖最大化以独特地使得能够进行成对3d基因组相互作用的最高分辨率和最灵敏的检测也适用于其它形式的hic和其衍生方案,特别是capture-hic、hichip、tcc以及其它基于限制性酶的全基因组测定或基于靶向hic的测定。
84.蛋白质因子定位
85.图9显示在超高re切割位点密度(hicoverage)中使基因组覆盖最大化如何独特地使得能够在hichip(plac-seq)测定中进行更灵敏的蛋白质因子定位分析和3d构象分析。在hichip测定中,剪切邻位连接的染色质并且使其富集所关注的蛋白质因子(ctcf、h3k27ac、
内聚亚基蛋白质、h3k4me3等)。在纯化蛋白质结合的连接dna之后,富集连接接点,产生包括由特定蛋白质-因子介导的连接事件的核酸模板。这样,hichip不仅提供关于蛋白质因子定位的信息(类似于chip-seq),而且提供关于3d基因组构象的信息(类似于hic)。一个主要的限制因素是,为了将核酸制备为模板,它必须与蛋白质因子定位位点及限制性酶切割位点两者线性邻近。在hicoverage中,增加的re切割位点密度导致核酸模板中所表示的蛋白质因子定位位点的更大百分比增加,以及源于蛋白质因子定位位点的更多独特的连接接点。当对这些核酸模板进行测序时,来源于自核酸模板的序列数据有助于更灵敏的蛋白质定位分析(例如1d“峰值识别”,诸如在chip-seq中)和更灵敏的3d相互作用分析(例如2d“峰值识别”,诸如在hic中)。
86.变体发现和单倍型定相
87.图10a和图10b显示在超高re切割密度(hicoverage)中使基因组覆盖最大化如何独特地使得能够进行高度灵敏且同时的变体发现和单倍型分型分析。图10a显示了此在变体发现和单倍型定相的背景下的影响,沿着基因组的区域描绘4种het.snv。het.snv由于它们紧邻re切割位点而获得序列覆盖。如果het.snv远离re切割位点,则它不接收序列覆盖,因此不能被发现。而且,只有具有由hic提供的远程链接信息的snv可以用于基于读段的单倍型定相。通过超高re切割位置密度的hicoverage覆盖均匀性使得4/4het.snv能够获得序列覆盖,从而使小变体发现灵敏度和单倍型定相灵敏度最大化。在图10b中,鸟枪法wgs覆盖均匀性不局限于邻近re切割位点的区域,因此也使得4/4het.snv能够获得序列覆盖,从而使小变体发现灵敏度最大化。然而,鸟枪法wgs不包括远程邻接性信息,因此0/4snv可以是单倍型的。注意,描绘了杂合snv以举例说明变体灵敏度概念,但还可以使用超高re切割密度(hicoverage)以最大灵敏度同时进行其它类型变体的发现和单倍型定相。
88.甲基化分析
89.在hicoverage方法中使基因组覆盖最大化独特地使得能够对dna甲基化进行高度灵敏的分析,并且可与传统的全基因组亚硫酸氢盐测序(wgbs)相当。只有邻近re切割位点的胞嘧啶将存在于核酸模板中并且可用于亚硫酸氢盐转化和甲基化状态的确定。远离re切割位点的胞嘧啶将是未知的,因为那些核酸将不存在于核酸模板中。通过超高re切割位点密度的hicoverage均匀性使得能够检测所有胞嘧啶的甲基化状态,这是因为它们相对于re切割位点邻近定位。其他类型的dna甲基化,例如羟甲基化胞嘧啶,也可以借助于基因组覆盖使用hicoverage灵敏地检测(将亚硫酸氢盐转化应用于一组模板并将tab-seq应用于另一组模板,并且使用两个数据集确定mc和hmc状态)。
90.在一些实施方案中,在pcr和测序之前,将通过本文所述方法生成的具有保持的空间邻近的邻接性信息的核酸与亚硫酸氢盐试剂接触,以使得能够在碱基分辨率下同时分析空间邻近性和dna甲基化。在一些实施方案中,亚硫酸氢盐试剂是亚硫酸氢钠。
91.在一些实施方案中,如先前所述(rao等人,cell,159(7),第1665-1680页(2014),li等人,nature methods,16(10),第991-993页(2018)),使用hic方案生成hic连接产物。使用链霉抗生物素蛋白珠粒来富集连接接点。如先前所述(rao等人,cell(2014)),接着进行illumina文库构建,同时将dna附接到链霉抗生物素蛋白珠粒。直接在接头(adapter)连接之后,使用本领域已知的方法,对dna进行亚硫酸氢盐转化。在亚硫酸氢盐转化之前以0.5%掺入未甲基化的λdna,以估计转化率。对亚硫酸氢盐转化的dna进行纯化、扩增和测序。
92.在一些实施方案中,将剪切的hic连接产物用亚硫酸氢盐试剂处理并纯化(stamenova等人,biorxiv,第481283页(2018))。然后使用链霉抗生物素蛋白珠粒来富集连接接点。然后从珠粒中分离dna,并使用本领域已知的用于将ssdna转化为dsdna测序文库的技术将所述dna制备为测序文库。然后对接头连接的分子进行文库扩增和测序。类似地,本领域已知的方法还可应用于分析dna甲基化状态(lister等人,nature,462(7271),第315-322页(2009);shultz等人,nature,523(7559),第212-21页(2015)))。此外,本领域已知的方法还可用于同时分析关于3d基因组折叠的dna甲基化状态(li等人,nature methods,16(10),第991-993页(2018);stamenova等人,biorxiv(2018)),并行揭示dna化学修饰性质和dna折叠模式。具体地说,在将该方法应用于蛋白质:cfdna复合物的情况下,本领域众所周知,无细胞核酸的dna甲基化状态可以告知组织来源分析以及几种其它cfdna分析,包括但不限于肿瘤dna的非侵入性检测、产前诊断和器官移植监测(zeng等人,journal of genetics and genomics,45(4),第185-192页(2018);lehmann-werman等人,proceedings of the national academy of sciences,113(13),第e1826-e1834页(2016))。
93.snv发现
94.使基因组覆盖(序列覆盖和均匀性)最大化能够实现高度灵敏的小变体灵敏度。snv由于其紧邻re切割位点而获得序列覆盖。因此,远离re切割位点的snv未接收到序列覆盖,因此不能被发现。在本文所述的方法中,通过超高re切割位点密度的覆盖均匀性使得基本上所有snv均能够获得序列覆盖,从而使小变体灵敏度最大化到如如用鸟枪法wgs所证实的等同水平。标准hic造成远离re切割位点的许多snv,因此无法发现。使用所述方法,可以以最大灵敏度发现许多类型的小变体,包括杂合snv(单核苷酸变体)、其它类型的snv和indel(插入和缺失)。
95.碱基校正
96.除了hic的已知基因组拼接(scaffolding)能力外,在hicoverage方法中使基因组覆盖最大化还独特地使得能够对最初通过易错测序技术检测到的错误基因组碱基进行高度灵敏的碱基校正,这可与鸟枪法wgs相当。在一种场景中,现行从头基因组组装工作流程通常涉及以下的组合:相对易错的长读测序技术(例如oxford nanopore,uk)以产生最连续的序列(“重叠群”),之后进行hic以将重叠群拼接转化为染色体规模的支架(scaffold),之后进行鸟枪法wgs(10x genomics,pleasanton,ca)以“校正”由易错的长读技术产生的错误碱基判读。由于核酸模板中的基因组表示不均匀,因而测序数据的覆盖不均匀,因此hic尚未被视为能够进行灵敏的碱基校正的技术。然而,使用通过超高re切割位置密度的hicoverage方法均匀性使得能够实现与鸟枪法wgs相当的最大碱基校正灵敏度。除了错误的单个碱基判读之外,通过易错测序技术产生的其它类型的错误dna序列也可以使用hicoverage方法,借助于均匀基因组覆盖来灵敏地校正。
97.cnv分析
98.在一些实施方案中,在hicoverage方法中使基因组覆盖最大化独特地使得能够进行与鸟枪法wgs相当的高灵敏cnv分析。cnv由于其与re切割位点的重叠而获得序列覆盖,而远离re切割位点的cnv未接收到序列覆盖,因此不能被发现或分析。hicoverage方法通过超高re切割位点密度提供覆盖均匀性,从而使cnv检测灵敏度最大化。使用所述超高re切割位点密度方法,可以以最大灵敏度发现和分析cnv,诸如扩增区域和杂合或纯合缺失。
99.数据分析/应用
100.下面表示一些数据分析和应用的采样,但并不意味着全部都包括在内。本领域已知将hic数据用于能够保持邻接性的分析和应用,诸如单倍型定相和基因组重排检测。例如,selvaraj等人,bmc genomics,16(1),第900页(2015),selvaraj等人,nature biotechnology,31(12),第1111页(2013)以及pct/us2014/047243描述了用于单倍型定相的hic数据,engreitz等人(plos one 2012年9月/第7卷/第9期/e44196)描述了用于人疾病中的基因组重排分析的hic数据。几篇其它文献已描述了使用hic数据进行基因组重排检测(dixon等人,nature genetics,50(10),第1388-1398页(2018);chakraborty和ay,bioinformatics(2018);harewood等人,genome biology,18(1),第125页(2017))。一种用于重排检测的此类分析工具是来自dixon等人,nature genetics(2018)的hic-breakfinder工具(https/github.com/dixonlab/hic_breakfinder)。其它能够保持邻接性的分析和应用包括但不限于从头基因组和宏基因组组装、结构变异检测等。
101.在测序之后,本领域已知的方法可用于在空间邻近性和远程序列邻接性的背景下分析数据,诸如但不限于使用空间邻近的邻接性信息来告知基因组折叠模式(lieberman-aiden等人,science,326(5950),第289-293页(2009))以及基因组重排分析(dixon等人,nature genetics,(2018))。
102.此外,由于已知hic信号独特地捕获远程序列邻接性信息以显著地增强基因组重排分析(dixon等人,nature genetics(2018)),因此应用于cfdna的hic可以富集来自液体活组织检查样品的此类基因组重排信号,并且极大地有利于早期非侵入性癌症诊断。最后,dna甲基化以及dna空间邻近性和远程邻接性两者的组合和同时分析将协同作用,以更好地实现本文所述的分析。
103.3c方法
104.在一些实施方案中,使用优化的基于3c的方法而不是hic方法来生成邻位连接产物。基于3c的方法包括但不限于3c、4c、5c、capture-c、3c-chip或methyl-3c。
105.在一些实施方案中,3c方法不像hic那样在连接接点中掺入标记或标记物。例如,生物素化的核苷酸或生物素化的桥接接头。
106.样品通常被交联以保持空间邻近的信息,然而可能并不始终需要样品的交联(bryant等人,mol syst biol.12(12):891(2016))。在一些实施方案中,本文所述的3c方法与组织、细胞、细胞核的样品一起使用,所述组织、细胞、细胞核不是交联的,但具有具稳定空间相互作用的空间邻近的dna分子。本文所述的适用于交联样品的3c方法的实施方案也旨在适用于未交联的样品。
107.本文所述的3c方法可在异位或原位进行。
108.在一些实施方案中,对3c方法进行优化以改善保持的空间邻近的邻接性信息量。远程顺式捕获的空间邻近的核酸(cspna)(在线性序列距离上大于15kb)对于邻接性应用是最具信息性的,并且通常用作用于确定空间邻近的邻接性信息的保持的替代物(proxy)。具体地说,用于测序的核酸模板中有多少百分比是远程顺式分子。在某些实施方案中,对3c方法进行优化以提高远程顺式分子的百分比。
109.在一些实施方案中,优化的3c方法还增加了含有连接接点的读段对的基因组覆盖均匀性。
110.在一些实施方案中,优化的3c基于使用多个限制性核酸内切酶(优化的3c邻位连接)(参见实施例4和实施例5以及图11和图12)。在一些实施方案中,优化的3c包括用于邻位连接分子的大小选择(参见实施例7和图13)以及多个限制性核酸内切酶的使用。
111.限制性核酸内切酶
112.在一些实施方案中,将样品的dna分子与两种或更多种限制性核酸内切酶、3种或更多种限制性核酸内切酶、4种或更多种限制性核酸内切酶、5种或更多种限制性核酸内切酶、6种或更多种限制性核酸内切酶、7种或更多种限制性核酸内切酶、8种或更多种限制性核酸内切酶、9种或更多种限制性核酸内切酶、10种或更多种限制性核酸内切酶、或更多种限制性核酸内切酶(例如2、3、4、5、6、7、8、9或10种限制性核酸内切酶)接触并消化。在某些实施方案中,一组限制性核酸内切酶是两种限制性核酸内切酶。在某些实施方案中,一组限制性核酸内切酶是三种限制性核酸内切酶。在某些实施方案中,一组限制性核酸内切酶是两种限制性核酸内切酶,并且所述限制性核酸内切酶中的一种是nlaiii。在一些实施方案中,限制性内切核酸酶中的一种是nlaiii,并且另一种限制性内切核酸酶是mboi或msei。在某些实施方案中,一组限制性核酸内切酶是三种限制性核酸内切酶,并且所述限制性核酸内切酶中的一种是nlaiii。在一些实施方案中,一组限制性核酸内切酶是三种限制性核酸内切酶,并且所述限制性核酸内切酶中的一种是nlaiii,且所述限制性核酸内切酶中的另一种是mboi或msei。在一些实施方案中,限制性核酸内切酶是nlaiii、mboi和msei。本文所述的方法涵盖增强空间邻近的邻接性信息保持的其它限制性核酸内切酶以及限制性核酸内切酶的组合。
113.在一些实施方案中,限制性酶导致相同的突出序列。此类酶的示例包括:acii、hinp1i、hpaii、hpych4iv、mspi和taqi-所有这些均在负dna链的5’末端上具有3
’‑
cg-5’突出端。类似地,bfai、msei和cviqi在负dna链的5’末端上具有3
’‑
ta-5’突出端。
114.在一些实施方案中,限制性酶导致不同的突出序列。
115.在一些实施方案中,dna分子与两种或更多种限制性核酸内切酶的接触和消化是一次性(即同时)进行。在某些实施方案中,然后使dna分子的所得空间邻近的消化末端与连接酶接触以生成连接接点。
116.在某些实施方案中,按顺序进行与两种或更多种限制性核酸内切酶的接触和消化。在一些实施方案中,每个顺序接触和消化事件可以用一种或多种限制性核酸内切酶进行。例如,接触和消化事件可以是用两种限制性核酸内切酶进行共消化。在一些实施方案中,基于所用的特定限制性核酸内切酶以限定的顺序进行用两种或更多种限制性核酸内切酶的顺序接触和消化。在某些实施方案中,在顺序消化(无论有序与否)结束时,使dna分子的所得空间邻近的消化末端与连接酶接触以生成连接接点。
117.在某些实施方案中,按顺序进行与每种限制性核酸内切酶或限制性核酸内切酶的组合的接触和消化,并且在通过一种或多种限制性核酸内切酶完成每个消化事件之后,使dna分子的所得空间邻近的消化末端与连接酶接触以生成连接接点(参见实施例8以及图16a和图16b)。用一种或多种不同的限制性核酸内切酶进行序列中的下一个消化事件,并且在消化结束后,使dna分子的空间邻近的消化末端与连接酶接触以生成进一步的连接接点。在一些实施方案中,顺序消化/连接可以重复2、3、4、5、6次或更多次。在某些实施方案中,多个限制性核酸内切酶消化/连接步骤基于所使用的特定限制性核酸内切酶以限定顺序实
施。
118.在某些实施方案中,优化的3c方法涵盖限制性核酸内切酶的其它组合、所产生的突出端的类型(相同、不同、或相同与不同的混合物)、同时或顺序消化、限制性核酸内切酶的顺序、每个顺序步骤中限制性核酸内切酶的数目以及是否进行在所有消化结束时或更频繁地在每次顺序消化之后进行一次连接,这改善了包括连接接点的分子的空间邻近的邻接性信息的保持和/或基因组覆盖。
119.大小选择
120.在一些实施方案中,使通过使用两种或更多种限制性核酸内切酶而产生的邻位连接dna分子富集含有保持空间邻近的邻接性的连接接点的分子。在某些实施方案中,通过大小选择进行富集。在一些实施方案中,大小选择是针对大小大约》5kb、》10kb、》20kb、》30kb、》40kb、》50kb或》60kb的较大片段。可以通过本领域已知的任何手段实施大小选择。
121.在一些实施方案中,直接在交联反转之后进行大小选择(如果使邻位连接的分子交联)。在某些实施方案中,可以通过使用手动或自动化方法(例如sage science bluepippin仪器(beverly,ma))或使用基于大小选择性dna沉淀的方法(例如circuloomics short read eliminator试剂盒(baltimore,md))进行凝胶提取,来进行大小选择。
122.在一些实施方案中,在邻位连接的分子片段化之后实施大小选择。在某些实施方案中,大小选择采用包被有非特异性地且可逆地结合dna的羧基的磁珠,例如固相可逆固定(spri)珠粒,如ampure beads(beckman coulter;brea,ca)。在某些实施方案中,可以调节珠粒与样品体积的比率以选择较大的片段。例如,比率可以是0.4
×
至0.8
×
或0.4
×
、0.5
×
、0.6
×
、0.7
×
、或0.8
×
。
123.在一些实施方案中,在文库制备期间,例如在进行pcr之前或之后,实施大小选择。可应用多种大小选择手段,包括使用spri珠粒。在构建文库之前进行的所述方法的大小选择并不涉及供特定测序机使用的特定大小的分子的优化。相反,如所述方法中所用的大小选择是针对通过影响含有连接接点的模板的比例并保持空间邻近的邻接性来增强数据组成的目的。例如,与对于优化的3c推荐的大约700bp的大得多的插入大小相比,对于hiseq仪器,最大平均文库插入大小推荐为350-450bp。
124.在一些实施方案中,优化的3c方案可以没有大小选择步骤,或者可以具有单个大小选择步骤、两个大小选择步骤或三个大小选择步骤。
125.在某些实施方案中,可以通过例如检查代表远程顺式分子的模板分子的百分比来评估用于大小选择的手段、所选择的大小范围以及使用超过一个大小选择步骤的适用性对改善空间邻近的邻接性信息的保持的作用。
126.通过利用多个限制性核酸内切酶或多个限制性核酸内切酶和大小选择,可以优化3c方法以改善空间邻近的邻接性信息的保持。多种限制性核酸内切酶消化的所述变化形式中的任一种可单独利用,或与大小选择的所述变化形式中的任一种组合利用。例如,在使用0.4
×
spri珠粒与样品体积的比率将邻位连接的分子片段化之后进行的非常严格的大小选择可与连续轮次的共消化和连接组合。
127.在一些实施方案中,如本文所述的优化的3c方法导致来源于基本上覆盖整个基因组的序列的邻位连接dna分子。
128.在一些实施方案中,从细胞核结构可保持完整的任何样品类型获得dna分子。在一
些实施方案中,从选自细胞核、细胞、组织、细胞系、原代细胞、解离组织、磨碎组织、福尔马林固定的石蜡包埋(ffpe)样品、ffpe组织切片或冷冻组织切片、深度福尔马林固定的样品或无细胞dna的样品中获得dna分子。在某些实施方案中,样品在水溶液中。在某些实施方案中,样品附着到诸如载玻片的固体表面。在某些实施方案中,样品在水溶液中。在一些实施方案中,在载玻片上分析ffpe组织。在一些实施方案中,分析从载玻片移除(例如,物理刮除、或通过使用激光捕获显微解剖)的ffpe组织。在一些实施方案中,在载玻片上分析冷冻组织。在一些实施方案中,分析从载玻片移除(例如,刮除)的冷冻组织。
129.在一些实施方案中,dna分子得自单个细胞,得自两个或更多个细胞,或得自组织样品或组织样品的特定部分。在一些实施方案中,样品的dna分子包括两个或更多个基因组或其部分。
130.在一些实施方案中,在制备用于测序的文库之前,纯化包括连接接点的邻位连接dna分子。在某些实施方案中,如果样品被交联,则使包括连接接点的邻位连接dna分子与逆转交联的试剂接触。
131.在一些实施方案中,由通过本文所述的优化的3c方法产生的邻位连接dna分子制备用于dna测序的模板分子文库。
132.在某些实施方案中,优化的3c方法包括一个或多个针对4c、5c、capture-c、3c-chip(3c邻位连接,之后chip-seq)或methyl-3c方法的步骤。
133.在一些实施方案中,由优化的3c方法的产物制备用于dna测序的模板分子文库,所述优化的3c方法包括以下中的一个或多个步骤:4c、5c、capture-c、3c-chip或methyl-3c方法。
134.在一些实施方案中,对模板分子文库进行测序以生成包括反映3c(3c-seq)的使用的序列信息的序列读段。在一些实施方案中,对模板分子文库进行测序以生成包括反映4c、5c、capture-c、3c-chip或methyl-3c方法的使用的序列信息的序列读段。
135.在某些实施方案中,测序是短读测序。在某些实施方案中,本文所述的优化的3c方法导致用于制备用于短读测序的文库的核酸模板的至少30%、至少40%、至少50%或至少60%是远程顺式分子。
136.在一些实施方案中,在制备用于短读测序的文库之前,将邻位连接dna分子片段化以生成邻位连接dna分子的片段,所述邻位连接dna分子的片段包括跨越连接接点的片段。
137.在某些实施方案中,测序是长读测序。
138.在一些实施方案中,对如本文所述通过利用优化的3c方案以及一个或多个针对4c、5c、capture-c、3c-chip或methyl-3c方法的步骤所制备的模板分子库进行测序以生成包括序列信息的序列读段。在某些实施方案中,测序是短读测序。在某些实施方案中,测序是长读测序。
139.文库制备、测序和序列信息的分析如本文前面所述。
140.在一些实施方案中,在分析空间邻近的邻接性的应用中利用序列信息。在某些实施方案中,序列信息用于检测基因组或其部分的成对3d基因组相互作用。在某些实施方案中,3d基因组相互作用在启动子、增强子、基因调控元件、gwas基因座、染色质环和拓扑结构域锚、重复元件、多梳区、基因体、外显子或整合的病毒序列之间。在某些实施方案中,序列信息用于基因组或其部分的蛋白质因子定位分析和3d构象分析。在某些实施方案中,蛋白
质因子定位分析和3d构象分析包括3c-chip。
141.在一些实施方案中,在受益于含有连接接点的读段对的增加的覆盖均匀性的应用中利用优化的3c方法。在某些实施方案中,序列信息用于基因组或其部分的重叠群的聚类和排序。在某些实施方案中,序列信息包括用于被聚类和排序的每个重叠群的序列信息。在某些实施方案中,序列信息用于对基因组或其部分的重叠群进行聚类、排序和定向。在一些实施方案中,序列信息用于基因组或其部分的单倍型定相。在一些实施方案中,序列信息用于宏基因组组装。
142.在一些实施方案中,在依赖于1d基因组覆盖的应用中利用序列信息。在某些实施方案中,序列信息用于基因组或其部分的基因组重排分析。在某些实施方案中,基因组重排分析包括断点的鉴别。在某些实施方案中,给定序列读段的序列信息位于断点的上游和下游。在某些实施方案,序列信息用于基因组或其部分的dna甲基化分析。在某些实施方案中,序列信息用于基因组或其部分的单核苷酸变体(snv)发现。在某些实施方案中,序列信息用于基因组或其部分的远程测序信息的碱基校正。在某些实施方案中,序列信息用于基因组或其部分的高灵敏拷贝数变异(cnv)分析。在某些实施方案中,拷贝数变异(cnv)是扩增。在某些实施方案中,拷贝数变异(cnv)是杂合或纯合缺失。
143.在某些实施方案中,序列信息用于基因组或其部分的变体发现、单倍型定相和基因组组装。在某些实施方案中,序列信息用于包括父本基因组的第一样品和包括母本基因组的第二样品中的变体发现和单倍型定相,并且父本基因组和母本基因组的定相变体用于分析从母体的cfdna中获得的胎儿基因组的序列数据。在某些实施方案中,序列信息用于基因组或其部分的单倍型定相和基因组组装。
144.在某些实施方案中,序列信息用于基因组或其部分的基因组组装和3d构象分析。在某些实施方案中,序列信息用于基因组或其部分的dna甲基化分析和3d基因组相互作用的检测。在某些实施方案中,序列信息用于基因组或其部分的基因组组装和3d基因组相互作用的检测。
145.在一些实施方案中,除了在连接接点中保持的空间邻近的邻接性信息之外,还保持了邻位连接dna分子的分子邻接性信息。在某些实施方案中,使用条形码来保持分子邻接性信息。在某些实施方案中,通过在文库制备之前使邻位连接dna与条形码化转座体连接珠粒接触,将条形码引入到邻位连接dna分子中。在某些实施方案中,通过利用所保持的邻位连接dna分子的分子邻接性,将序列信息用于检测基因组或其部分的更高级3d基因组相互作用。在某些实施方案中,通过利用所保持的邻位连接dna分子的分子邻接性,将序列信息用于检测基因组或其部分的三种或更多种同时进行的3d基因组相互作用。在某些实施方案中,通过利用所保持的邻位连接dna分子的分子邻接性,将序列信息用于检测虚拟的成对3d基因组相互作用。在某些实施方案中,虚拟的成对3d基因组相互作用在基因组或其部分的给定邻位连接dna分子内彼此不直接连接的限制性片段之间。
146.在某些实施方案中,通过利用所保持的邻位连接dna分子的分子邻接性获得的成对相互作用、虚拟的成对相互作用、和/或更高级相互作用用于基因组或其部分的3d基因组相互作用、基因组或其部分的基因组重排分析、基因组或其部分的重叠群的聚类和排序、确定基因组或其部分的重叠群定向、基因组或其部分的单倍型定相、基因组或其部分的dna甲基化分析、基因组或其部分的单核苷酸变体(snv)发现、基因组或其部分的远程测序信息的
碱基校正、基因组或其部分的高灵敏拷贝数变异(cnv)分析或其组合。
147.单细胞
148.在一些实施方案中,优化的3c方案是从提供单个细胞谱(profile)的单个细胞中获得序列信息。
149.通过细胞/细胞核分选(在3c之前或之后)的单细胞3c(“平板(plate)”方法)
150.在一些实施方案中,“大批量(in bulk)”(即在细胞群体中)实施原位3c邻位连接。使用细胞分选仪器(例如facs和fans)或手动将细胞/细胞核分选到离散的物理隔室(诸如微量滴定板的孔)中。使用本领域已知的全基因组扩增方法,诸如多重置换扩增(mda)或其它手段,从每一单个细胞纯化和扩增dna。此类方法类似于flyamer等人,nature,544(7648),第110-114页(2017)或tan等人,science,361(6405),第924-928页(2018)。由每个细胞/细胞核的扩增dna分子产生文库。对文库进行测序并检查序列读段以获得单个细胞分辨率的序列信息。
151.在一些实施方案中,可以通过保持来自每个单个细胞的每个邻位连接dna分子的分子邻接性来捕获每个细胞更多的成对相互作用。在某些实施方案中,将条形码化转座体连接的珠粒(例如tell-seq珠粒,universal sequencing technologies,carlsbad,ca)应用于每个微孔中纯化的邻位连接dna。在应用转座体连接的珠粒后,为每个单独的细胞构建文库。使用“虚拟对”的概念,来自每个单个细胞的邻位连接dna分子的重建具有显著改善每个细胞成对接触的数目的潜力,这意味着在连接产物中连接在一起的10个限制性片段通常将来源于大约9个连接接点,并产生9个成对3d接触。如果揭示给定连接产物上的全部10个片段,则这将告知成对3d接触的45个总组合((10*9)/2),或公式p=(((n*(n-1))/2),其中p是每个连接产物获得的成对3d接触的总数,并且n是串联到连接产物中的限制性片段的数目。如果连接产物中有25个限制性片段,则这在传统文库制备情况下将产生约24个成对接触,或者如果在文库制备期间保持每种3c连接产物的分子邻接性,则将产生300个“虚拟对”。这将表示每个细胞的信息内容的对数级增加。
152.通过微滴微流控方法(“微滴”方法)的单细胞3c
153.在一些实施方案中,“大批量”(即在细胞群体中)实施原位3c邻位连接。将细胞/细胞核输入商业的(例如,10
×
genomics(pleasanton,ca)、bio-rad(hercules,ca)、mission bio(south san francisco,ca)或自制的(例如drop-seq)微滴微流控系统中,其中将试剂递送到条形码并扩增来自每个单个细胞/细胞核的邻位连接的dna。由每个细胞/细胞核的扩增dna分子产生文库。对文库进行测序并检查序列读段以获得单个细胞分辨率的序列信息。
154.在一些实施方案中,4c用于文库制备(单细胞4c)。对于平板和微滴单个细胞方法中的4c,用包括细胞条形码而不是全基因组扩增的基因座特异性引物对(这是在4c中完成的)实施靶向扩增。
155.在一些实施方案中,将capture c用于富集特定靶标(通过靶富集来富集模板并对其进行测序)。由于模板具有基于用于获得单个细胞的方案的一个或多个细胞条形码(参见上文),因此可以将序列信息分配给单个细胞。
156.空间定位(“空间”方法)
157.在一些实施方案中,使用优化的3c方案(或hic方案)处理的组织切片的分析可以
为从组织切片的部分或从单个细胞中获得的序列信息提供空间定位。在某些实施方案中,在诸如载玻片的表面上完整保持组织的同时,实施原位3c(或hic)邻位连接,然后将组织(现包含邻位连接的细胞核)显微解剖成空间上不同的区域。在一些实施方案中,空间上不同的区域为网格(例如,8
×
12),其有时具有象限、同心圆(像牛眼)、接触非肿瘤细胞或肿瘤微环境的外周肿瘤细胞、组织亚区域中的细胞簇、或单个细胞的集合。可将每个空间上不同的区域视为自身的“样品”,并作为不同的细胞物理集合进行处理,或者可以根据上述示例获得单个细胞并单独处理。在某些实施方案中,首先将组织切片显微解剖成空间上不同的区域,并将每个空间上不同的区域视为其自身的原位3c(或hic)邻位连接反应,并作为不同的细胞物理集合进行处理,或者可以根据上述示例获得单个细胞并单独处理。在数据分析阶段期间,空间上不同区域的组织3c(或hic)谱或单个细胞3c(或hic)谱可归因于它们在组织切片内的空间定位。
158.在某些实施方案中,每个空间上不同的区域可能不需要像其自身单独的原位3c(或hic)反应那样被处理。在某些实施方案中,类似于multi-seq(mcginnis等人,nature methods,16(7),第619页(2019))的方法可以被修改以用于单个细胞3c(或hic)分析背景中对样品条形编码。例如,可以从来自组织切片的每个空间限定的区域收集细胞/细胞核。然后将使样品与脂质修饰的寡核苷酸(lmo)或胆固醇修饰的寡核苷酸(cmo)反应,所述寡核苷酸嵌入细胞膜的质膜或核膜中。寡核苷酸将包括在邻位连接的细胞核被分配到平板的孔或微滴中之后被扩增的手段。在数据分析阶段期间,单个细胞3c(或hic)谱可归因于它们在组织切片内的空间定位,并且对应于每个单个细胞的共扩增的样品条形码序列将用作在样品标记反应期间引入的样品标识符。
159.在一些实施方案中,在组织切片的分析中利用4c。使用3c模板用基因座特异性引物对实施靶向扩增,所述3c模板由从组织切片显微解剖的每个空间限定的区域产生。
160.文库制备操作
161.在一些实施方案中,将上述3c方法与靶富集方法组合。在某些实施方案中,靶富集基于pcr。
162.文库制备后操作
163.在一些实施方案中,将上述3c方法与靶富集方法组合。在某些实施方案中,靶富集基于探针。在某些实施方案中,靶富集基于pcr。
164.在一些实施方案中,将capture c用于富集特定靶标(通过靶富集来富集模板并对其进行测序)。
165.试剂盒
166.在一些实施方案中,提供了用于实施本文所述方法的试剂盒。试剂盒通常包括一个或多个容器,所述容器含有本文所述的一种或多种部件。试剂盒包括任何数目的单独容器、小包(packet)、管、小瓶、多孔板等中的一个或多个部件,或者部件可以在此类容器中以各种组合的形式进行组合。试剂盒部件和试剂如本文所述。
167.hic试剂盒
168.在一些实施方案中,试剂盒包括以下中的一种或多种:(a)三种或更多种限制性核酸内切酶;
169.(b)限制性核酸内切酶缓冲液;以及(c)以下中的一种或多种:生物素化的核苷酸、
未标记的核苷酸、dna聚合酶、连接酶、连接酶缓冲液、一种或多种附加的用于逆转交联的缓冲液和试剂。
170.在一些实施方案中,试剂盒包括以下中的一种或多种:(a)四种限制性核酸内切酶;(b)限制性核酸内切酶缓冲液;以及(c)以下中的一种或多种:生物素化的核苷酸、未标记的核苷酸、dna聚合酶、连接酶、连接酶缓冲液、一种或多种附加的用于逆转交联的缓冲液和试剂。在某些实施方案中,四种限制性核酸内切酶是:mboi、hinfi、msei和ddei。在某些实施方案中,四种限制性核酸内切酶是:hpych4iv、hinfi、hinp1i和msei。
171.在一些实施方案中,试剂盒包括以下中的一种或多种:四种限制性核酸内切酶;
172.(b)两种或更多种限制性核酸内切酶缓冲液;以及(c)以下中的一种或多种:生物素化的核苷酸、未标记的核苷酸、dna聚合酶、连接酶、连接酶缓冲液、一种或多种附加的用于逆转交联的缓冲液和试剂。在一些实施方案中,两种或更多种限制核酸内切酶缓冲液在与四种限制性核酸内切酶分开的容器中。在一些实施方案中,每种限制性核酸内切酶具有至少1/256的理论消化频率。在一些实施方案中,至少两种限制性核酸内切酶需要用于高水平活性的独特缓冲液。
173.在一些实施方案中,限制性核酸内切酶在分开的容器中。在一些实施方案中,限制性核酸内切酶在单个容器中。在一些实施方案中,每种限制性核酸内切酶在常用的限制性核酸内切酶缓冲液中具有高活性水平,并且每种限制性核酸内切酶具有至少1/256的理论消化频率。在一些实施方案中,限制性核酸内切酶缓冲液在与限制性核酸内切酶分开的容器中。
174.3c试剂盒
175.在一些实施方案中,试剂盒包括以下中的一种或多种:(a)两种或更多种限制性核酸内切酶;
176.(b)限制性核酸内切酶缓冲液;以及(c)以下中的一种或多种:连接酶、连接酶缓冲液、一种或多种附加的用于逆转交联的缓冲液和试剂、一种或多种附加的用于大小选择的缓冲液和试剂、珠粒连接的转座体、具有条形码寡核苷酸的引物、一种或多种用于产生测序文库的试剂,并且不包括生物素化的核苷酸或标记的核苷酸。
177.在一些实施方案中,试剂盒包括以下中的一种或多种:(a)两种限制性核酸内切酶;(b)限制性核酸内切酶缓冲液;以及(c)以下中的一种或多种:连接酶、连接酶缓冲液、一种或多种附加的用于逆转交联的缓冲液和试剂、一种或多种附加的用于大小选择的缓冲液和试剂、珠粒连接的转座体、具有条形码寡核苷酸的引物、一种或多种用于产生测序文库的试剂,并且不包括生物素化的核苷酸或标记的核苷酸。在某些实施方案中,限制性核酸内切酶中的一种是nlaiii。在某些实施方案中,限制性内切核酸酶中的一种是nlaiii,并且另一种限制性内切核酸酶是mboi或msei。
178.在一些实施方案中,试剂盒包括以下中的一种或多种:(a)三种限制性核酸内切酶;
179.(b)限制性核酸内切酶缓冲液中的一种或多种;以及(c)以下中的一种或多种:连接酶、连接酶缓冲液、一种或多种附加的用于逆转交联的缓冲液和试剂、一种或多种附加的用于大小选择的缓冲液和试剂、珠粒连接的转座体、具有条形码寡核苷酸的引物、一种或多种用于产生测序文库的试剂,并且不包括生物素化的核苷酸或标记的核苷酸。在某些实施
方案中,限制性核酸内切酶中的一种是nlaiii。在某些实施方案中,限制性内切核酸酶中的一种是nlaiii,且其它限制性内切核酸酶中的一种是mboi或msei。在某些实施方案中,限制性核酸内切酶是:nlaiii、mboi和msei。
180.在一些实施方案中,试剂盒的限制性核酸内切酶产生相同的突出序列。在一些实施方案中,试剂盒的限制性核酸内切酶产生不同的突出序列。在一些实施方案中,可以同时用试剂盒的两种或更多种限制性核酸内切酶实施消化。在一些实施方案中,不能同时用试剂盒的两种或更多种限制性核酸内切酶实施消化。
181.在一些实施方案中,试剂盒的限制性核酸内切酶在分开的容器中。在一些实施方案中,试剂盒的限制性核酸内切酶在单个容器中。在一些实施方案中,试剂盒的限制性核酸内切酶在超过一个容器中,并且至少一个容器装有超过一种限制性核酸内切酶。在一些实施方案中,试剂盒的每种限制性核酸内切酶在常用的限制性核酸内切酶缓冲液中具有高活性水平,并且缓冲液在一个容器中。在一些实施方案中,超过一种缓冲液在试剂盒中,并且缓冲液在分开的容器中。在一些实施方案中,限制性核酸内切酶缓冲液在与限制性核酸内切酶分开的容器中。
182.在某些实施方案中,试剂盒包括说明书。在一些实施方案中,说明书列举了试剂盒的限制性酶待使用的顺序。
183.试剂盒有时与方法结合使用,并且可以包括用于进行一种或多种方法的说明书和/或一种或多种组合物的描述。试剂盒可用于实施本文所述的方法。说明书和/或描述可以呈有形形式(例如纸等)或电子形式(例如缠结介质(例如光盘)等上的计算机可读文件),并且可以包括在试剂盒插页(insert)中。试剂盒还可以包括提供此类说明书或描述的因特网位置的书面描述。
184.文库
185.在一些实施方案中,基于使用hic或优化的3c方法,如本文所述构建文库。
186.实施例
187.以下所述的实施例说明某些实施方案,且并不限制本技术。
188.实施例1:最佳re的选择
189.图3a至图3b显示可与mboi结合使用以增加re切割位点密度和基因组覆盖的候选re的染色质消化效率。选择准则包括re必须在常用的re消化缓冲液中具有100%的活性水平。re还必须以足够高的浓度商购,使得在hic期间可以利用合理体积的每种酶。最后,re的组合必须使计算机中的消化频率(每种酶具有至少1/256的理论消化频率)最大化。这些准则将有助于确保re组合在hic背景下的生物化学相容性、效率和实用性,并且提供最大的基因组覆盖。
190.用增加量的hinfi以一式两份消化交联的gm19240细胞30min。在消化后,逆转交联,纯化dna,并进行凝胶电泳。有效的染色质消化需要至少100u的hinfi,这由消化的dna样品的较小分子量加以证明。由于hinfi可以用合理量的re单位(例如100单位)达到交联染色质消化的效率水平,并且与mboi相同的缓冲液相容,因此hinfi可以与mboi结合使用(参见图3a)。mboi和hinf1两者在cutsmart
tm
缓冲液(new england biolabs,beverly,ma)(1
×‑
50mm乙酸钾、20mm tris乙酸盐、10mm乙酸镁、100ug/ml bsa,ph 7.9,25℃下)中均有效地切割。
191.为了选择另外的re以进一步增加覆盖均匀性,鉴别出在也与mboi和hinfi相容的re缓冲液(cutsmart
tm
缓冲液)中具有100%报告活性水平的4种另外的4-切割酶(bfai、ddei、msei和mspi)。用最大实际量的每种酶以一式两份消化交联的gm12878细胞。在消化后,逆转交联,纯化dna,并进行凝胶电泳。令人惊讶的是,尽管re浓度、缓冲液相容性和计算机中的切割位点频率(1/256)合理,但在hic期间只有2/4的re显示有效的re消化(参见图3b)。选择这2种re ddei(至少25个单位)和msei(至少125个单位)作为待与mboi(至少100个单位)和hinfi(至少100个单位)结合使用的re,以达到最佳的re切割位点密度和基因组覆盖。然而,当用这四种酶同时消化交联的gm 12878细胞并通过凝胶电泳检查消化后片段的大小时,令人惊讶的是,消化后片段的大小与单一酶的大小相当(数据未显示)。这表明,即使使用四种酶的组合,也不是每个切割位点都被切割,并且不能预测可以获得与每个切割位点相邻的序列覆盖以实现完全基因组覆盖。
192.实施例2:snv发现
193.图4显示来自hicoverage的改善的基因组覆盖如何使得能够进行高度灵敏的snv发现,并且可与鸟枪法wgs相当。对于该分析,使用具有默认参数且包括-sp5m选项的bwa mem,将原始的2
×
150bp hic原始读段与hg19人基因组进行比对,该bwa mem将读段对作为单个末端进行比对,但保留配偶对信息,并且还保留最靠近5’的比对(5’most alignment)作为用于嵌合读段的主要比对。在比对后,使用gatk添加读取组(read group),并且使用picardtools去除pcr重复序列。然后将gatk用于base recalibration和print reads,然后使用gatk haplotype caller来判读变体,并使用gatk variant recalibration,以99.9的非默认份额值(tranche value)和4或8的maxgaussian设置进行重新校准。对于鸟枪法wgs数据,我们从bottle联盟(consortium)中的基因组中获得na12878、na24385和na24631的原始序列数据(zook,scientific data,2016)。对于na12878和na24385,对原始的2
×
148bp读段对进行二次采样,使得总深度可与供体匹配的hicoverage数据集相当。对于na24631,下载全部可用的2
×
250bp数据集且将其用于随后的分析。对于第4位个体(na19240),从steinberg等人,biorxiv,第067447页(2016)下载鸟枪法wgs数据,并进行二次采样,使得总深度可与供体匹配的hicoverage数据集相当。在如上文所述对数据集进行收集和采样之后,如上文所述对hic的读段对进行处理,除了在比对期间,将数据映射为真配偶对(-m),并且始终使用默认份额值(99.0%)和默认maxgaussian(8)来重新校准变体判读。对于所有hicoverage数据集,仅保留常染色体上支持性读取深度最小为5个读段的双等位基因纯合或杂合snv,用于相对于来自鸟枪法wgs数据的snv判读的“真实(truth)”集的snv灵敏度基准分析。对于giab基因组(na12878、na24385、na24631),我们进一步将用于基准分析的变体子集化为仅在由giab限定的高置信度区域中的那些变体。对于三个giab基因组中来自鸟枪法wgs数据的变体的真实集,我们使用常染色体上从giab联盟提出的相同高置信度区域提取的双等位基因纯合或杂合snv(zook,scientific data,2016)。对于na19240,从1000个基因组项目中获得真实变体判读。
194.实施例3:各种基因组的hicoverage
195.从各种来源(诸如用于脊椎动物的genomeark(https://vgp.github.io/genomeark/)和用于其他基因组的ncbi(https://www.ncbi.nlm.nih.gov/genome/))下载20种脊椎动物基因组组装体、两种植物基因组组装体、两种昆虫基因组组装体和两种寄生
虫基因组组装体。然后使用用于mboi、msei、ddei和hinfi的四种限制性酶切割位点基序,或者仅使用单一限制性酶mboi模拟相对低密度的限制性酶方法,在计算机中消化基因组。为了估计预期的覆盖,或将对hic“可见”的基因组碱基的分数,计算离限制性酶切割位点250bp以内的基因组碱基的分数。将这些分数针对每个基因组(x轴标记)(图14a
–
脊椎动物基因组;图14b
–
昆虫基因组、植物基因组和寄生虫基因组)标绘在y轴上。
196.结果表明,使用限制性酶组合的hicoverage使得能够进行跨代表性植物和动物物种的接近完全的基因组覆盖,因此各种植物和动物物种应当对本文所述hicoverage数据的独特益处稳健。
197.实施例4:hicoverage和覆盖均匀性
198.使用mboi、msei、ddei和hinfi对交联的gm12878细胞进行hicoverage实验,并测序到大约37
×
原始深度。从rao,cell,2014下载在gm12878细胞中使用mboi的深度匹配的低密度hic数据。使用bwa mem-sp5m将每个数据集映射到hg19参考基因组,并使用picardtools进行去重复。然后使用deeptools生成基因组覆盖直方图。如图15所示,结果显示观察到的覆盖均匀性的巨大差异,hicoverage数据的覆盖均匀性相对于低密度re方法显著改善。
199.实施例5:多酶3c
–
同时消化
200.使用mboi、nlaiii或msei,用一种、两种或三种限制性酶(在图11的分类轴标记上表示)以一式两份同时消化交联的gm12878细胞。消化后,使用连接酶进行邻位连接。然后,逆转交联并纯化邻位连接的dna。然后剪切邻位连接的dna,并使用0.6
×
的ampure beads与样品体积之比进行大小选择。最后,构建lllumina测序文库,进行pcr扩增,并使用0.6
×
的ampure beads与样品体积之比进行纯化。在miniseq上对3c文库进行测序,得到每个样品约1m的原始pe读段。在映射和去重复后,对于限制性酶共消化条件的每一排列,对代表远程(》15kb插入大小)染色体内相互作用的读段对的分数进行计数,并沿y轴标绘(参见图11)。
201.图11所示的测序结果表明,某些限制性酶的实施改善了核酸模板中空间邻近的邻接性的保持(当用于大小选择的背景下时)。所测试的限制性酶的最佳结果来源于包括nlaiii的条件。其次,使用两种限制性酶相对于使用单一酶(例如,nlaiii+mboi或msei相对于单独的nlaiiil)改善了核酸模板中空间邻近的邻接性的保持。然而,在这些特定条件(例如,nlaiii+mboi+msei或nlaiii+msei+mboi)下向混合剂中添加第三种酶并不进一步改善核酸模板中空间邻近的邻接性的保持,然而可以增加含有连接接点的核酸模板的覆盖均匀性。
202.实施例6:多酶3c
–
顺序消化
203.使用mboi、nlaiii或msei,用一种、两种或三种限制性酶以一式两份按顺序消化交联的gm12878细胞。将限制性酶消化的顺序表示为分类轴标记(参见图12)。例如,在三重消化(图12的条形图中的最右条柱)的情况下,首先用nlaiii消化gm12878细胞核。在nlaiii反应完成后,然后用mboi消化细胞核。在mboi消化完成后,然后用msei消化细胞核。在msei消化完成后,使用连接酶实施邻位连接。然后,逆转交联并纯化邻位连接的dna。然后剪切邻位连接的dna,并使用0.6
×
的ampure beads与样品体积之比进行大小选择。最后,构建lllumina测序文库,进行pcr扩增,并使用0.6
×
的ampure beads与样品体积之比进行纯化。在miniseq上对3c文库进行测序,得到每个样品约1m的原始pe读段。在映射和去重复后,对于每一条件,对代表远程(》15kb插入)染色体内相互作用的读段对分数进行计数,并沿y轴
标绘(参见图12)。
204.测序结果表明,相对于用单一酶消化相比,用》1种限制性酶消化样品改善了核酸模板中空间邻近的邻接性的保持。这一结果令人惊讶,因为用多种限制性酶消化产生了用于邻位连接的不相容末端,但邻位连接仍然通过远程顺式读出的分数的增加而得以证明。例如,以任一顺序用nlaiii和msei进行顺序消化,最大程度地改善了核酸模板中空间邻近的邻接性的保持。测序结果还表明顺序消化的顺序似乎影响测序结果(例如,从msei开始、之后是nlaiii的条件在核酸模板中具有最大的空间邻近的邻接性的保持)。然而,与共消化结果(图11)类似,在这些条件下,将第三种酶添加到一系列限制性消化中,相对于两次消化,不会进一步改善核酸模板中空间邻近的邻接性的保持,但可以增加含有连接接点的核酸模板的覆盖均匀性。不受理论约束,在限制性核酸内切酶的这一特定组合中的第三种酶未能增加核酸模板中空间邻近的邻接性的保持可能是因为不能邻位连接的不相容末端的增加。作为克服该问题的可能手段,可以使用产生相同突出序列且因此适用于3c实验中的粘性末端连接的限制性酶。克服该问题的另一种可能手段可以是进行顺序轮次的消化和连接。
205.实施例7:3c文库的大小选择
206.用nlaiii消化交联的gm12878细胞。消化后,使用连接酶进行邻位连接。然后,逆转交联并纯化邻位连接的dna。然后剪切邻位连接的dna并将其分成3组dna,并且使用0.7
×
、0.6
×
或0.5
×
的ampure beads与样品体积之比,以一式四份进行dna大小选择。使用12个dna样品构建illumina测序文库并进行pcr扩增。在pcr扩增后,将来自每组的2个文库使用0.6
×
的ampure beads与样品体积之比进行纯化,每组的另外2个文库使用0.8
×
的ampure beads与样品体积之比进行纯化(并进行大小选择)。在miniseq上对3c文库进行测序,得到每个样品约1m的原始pe读段。在映射和去重复后,对于剪切后和pcr后大小选择条件的每一排列,对代表远程(》15kb插入大小)染色体内相互作用的读段对分数进行计数,并沿y轴标绘。
207.图13所示的测序结果表明以下总体趋势:已经历偏向更大核酸模板(即,最小的ampure beads与样品体积的比率,条形图的右侧)的大小选择的文库显示核酸模板中空间邻近的邻接性的最大保持。例如,当仅考虑接收0.8
×
pcr后大小选择的条件时,远程顺式读出的分数从33%增加到36.5%、增加到39%。这是因为0.8
×
不大可能具有大小选择作用,因为它比最低剪切后大小选择的比率高,意味着剪切后大小选择参数(以及因此核酸模板的分子大小)在驱动测序结果。
208.实施例8:多酶3c
–
顺序轮次的消化和连接
209.对交联的gm12878细胞进行两轮连续的消化和邻位连接反应。在第一轮中,用mboi消化gm12878细胞核,然后使用连接酶进行邻位连接。然后将细胞核沉淀并且再悬浮在1
×
限制性消化缓冲液(cutsmart)中。然后使用nlaiii对细胞核进行第二轮限制性消化,然后用连接酶进行第二轮邻位连接。作为对照,在第一轮消化和邻位连接之后留出一些细胞核。然后,在所有的细胞核样品中逆转交联,并纯化邻位连接的dna。然后剪切邻位连接的dna,并使用0.7
×
的ampure beads与样品体积之比进行大小选择。最后,构建illumina测序文库,进行pcr扩增,并使用0.8
×
的ampure beads与样品体积之比进行纯化。在miniseq上对3c文库进行测序,得到每个样品约1m的原始pe读段。在映射和去重复后,对于每一条件,对
代表远程(》15kb插入)染色体内相互作用的读段对分数进行计数,并沿y轴标绘。在整个实验中,每次消化和连接反应后取细胞核的小等分试样(总共4等分试样),以在每个步骤后获得dna的分子大小。dna是通过交联逆转和dna纯化获得的这些细胞核等分试样。然后使用具有所示分子量梯度(ladder)的flashgel(lonza),通过凝胶电泳分析dna。
210.图16a显示凝胶电泳结果表明染色质正被mboi有效消化和再连接,这由消化的染色质的较低分子量和邻位连接后分子量的增加加以证明。结果还表明,邻位连接的染色质正被nlaiii有效地再消化和再连接,这由再消化的染色质的较低分子量和第二轮邻位连接后分子量的增加加以证明。测序结果表明,添加第二轮顺序消化和再连接可以改善核酸模板中空间邻近的邻接性的保持(参见图16b),同时增加含有连接接点的核酸模板的覆盖均匀性。
211.实施例9:实施方案的非限制性实施例
212.a1.一种用于从样品制备dna分子的方法,其包括:
213.(a)使包括基因组或其部分的样品的交联dna分子与一组限制性核酸内切酶接触;从而生成交联dna分子的空间邻近的消化末端;
214.(b)使交联dna分子的所述空间邻近的消化末端与连接酶接触,从而生成包括连接接点的交联的邻位连接dna分子;
215.(c)使所述交联的邻位连接dna分子与逆转交联的试剂接触,从而生成包括连接接点的邻位连接dna分子;以及
216.(d)将所述邻位连接dna分子片段化,以生成邻位连接dna分子片段,所述邻位连接dna分子片段包括跨越所述连接接点的片段,其中跨越所述连接接点且长度可为用于短程测序的模板的片段包括基本上全基因组或其部分的序列。
217.a2.根据实施方案a1所述的方法,其中跨越所述连接接点的所述片段包括多达750个碱基对的片段。
218.a3.根据实施方案a1或a2所述的方法,其中所述组的每种限制性核酸内切酶在常用缓冲液中具有高活性水平,并且所述组的每种限制性核酸内切酶具有至少1/256的理论消化频率。
219.a4.根据实施方案a1至a3中任一项所述的方法,其中所述组的限制性核酸内切酶由四种限制性核酸内切酶组成。
220.a5.根据实施方案a4所述的方法,其中所述限制性核酸内切酶是:mboi、hinfi、msei和ddei。
221.a5.1.根据实施方案a4所述的方法,其中所述限制性核酸内切酶是:hpych4iv、hinfi、hinp1i和msei。
222.a6.根据实施方案a1至a5.1中任一项所述的方法,其中从选自细胞核、细胞、组织、福尔马林固定的石蜡包埋(ffpe)样品、深度福尔马林固定的样品或无细胞dna的样品中获得所述dna分子。
223.a7.根据实施方案a1至a5.1中任一项所述的方法,其中从单个细胞中获得所述dna分子。
224.a7.1.根据实施方案a1至a5.1中任一项所述的方法,其中从两个或更多个细胞中获得所述dna分子。
225.a8.根据实施方案a1至a5.1中任一项所述的方法,其中样品的所述交联dna分子包括两个或更多个基因组或其部分。
226.a9.根据实施方案a1至a8中任一项所述的方法,其中在染色质构象测定中分析所述邻位连接dna分子。
227.a10.根据实施方案a9所述的方法,其中所述染色质构象测定是capture-c、3c、4c、5c、hic、capture-hic、hichip、plac-seq、栓系染色体捕获(tcc)、hiculfite、methyl-hic、hichirp或其组合。
228.a11.根据实施方案a9所述的方法,其中所述测定是全基因组的。
229.a11.1.根据实施方案a11所述的方法,其中所述测定是3c、hic、栓系染色体捕获(tcc)、hiculfite、methyl-hic或其组合。
230.a12.根据实施方案a9所述的方法,其中所述测定针对所述基因组中的一个或多个靶区域。
231.a12.1.根据实施方案a12所述的方法,其中所述测定是capture-c、4c、5c、capture-hic、hichip、plac-seq、hichlrp或其组合。
232.a13.根据实施方案a12所述的方法,其中所述靶标是单核苷酸变异、插入、缺失、拷贝数变异、基因组重排或用于定相的靶标。
233.a14.根据实施方案a12或a13所述的方法,其中所述样品包括癌症基因组,并且所述靶区域与表型相关。
234.a15.根据实施方案a1至a14中任一项所述的方法,其中使用所述邻位连接dna分子的片段来制备用于dna测序的模板分子文库,所述邻位连接dna分子的片段包括跨越所述连接接点的片段。
235.a15.1.根据实施方案a15所述的方法,其中用亲和纯化标记物标记所述连接接点。
236.a15.2根据实施方案a15.1所述的方法,其中所述亲和纯化标记物是与核苷酸缀合的生物素。
237.a15.3.根据实施方案a15.2所述的方法,其中通过用亲和纯化分子对所述亲和纯化标记物的亲和纯化进行富集。
238.a16.根据实施方案a15.3所述的方法,其中跨越所述连接接点的片段被富集以制备用于dna测序的模板分子文库。
239.a17.根据实施方案a15至a16中任一项所述的方法,其用于hic、capture-hic、hiscip、plac-seq、hiculfite或methyl-hic方法中。
240.a17.1.根据实施方案a15.3所述的方法,其中所述亲和纯化分子是链霉抗生物素蛋白。
241.a17.2.根据实施方案a16所述的方法,其中通过大小选择对包括连接接点的片段化的邻位连接dna分子进行富集。
242.a18.根据实施方案a15至a17.2中任一项所述的方法,其中所述模板分子文库提供基因组或其部分的均匀的全基因组覆盖。
243.a18.1.根据实施方案a15至a18中任一项所述的方法,其中对所述模板分子文库进行测序以生成包括序列信息的序列读段。
244.a19.根据实施方案a18.1所述的方法,其中所述测序是短读测序。
245.a20.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于所述基因组或其部分的基因组重排分析。
246.a21.根据实施方案a20所述的方法,其中所述基因组重排分析包括断点的鉴别。
247.a22.根据实施方案a21所述的方法,其中给定序列读段的序列信息位于所述断点的上游和下游。
248.a23.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于所述基因组或其部分的重叠群的聚类和排序。
249.a24.根据实施方案a23所述的方法,其中序列信息包括被聚类和排序的每个重叠群的序列信息。
250.a25.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于确定所述基因组或其部分的重叠群定向。
251.a26.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于对所述基因组或其部分的重叠群进行聚类、排序和定向。
252.a27.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于检测所述基因组或其部分的成对3d基因组相互作用。
253.a28.根据实施方案a27所述的方法,其中所述3d基因组相互作用在启动子、增强子、基因调控元件、gwas基因座、染色质环和拓扑结构域锚、重复元件、多梳区、基因体、外显子或整合的病毒序列之间。
254.a29.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于所述基因组或其部分的蛋白质因子定位分析和3d构象分析。
255.a30.根据实施方案a29所述的方法,其中所述蛋白质因子定位分析和3d构象分析包括plac-seq或hichip。
256.a31.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于所述基因组或其部分的单倍型定相。
257.a32.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于所述基因组或其部分的基因组组装和3d构象分析。
258.a33.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于所述基因组或其部分的dna甲基化分析。
259.a33.1.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于所述基因组或其部分的dna甲基化分析和3d基因组相互作用的检测。
260.a34.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于所述基因组或其部分的单核苷酸变体(snv)发现。
261.a35.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于所述基因组或其部分的远程测序信息的碱基校正。
262.a36.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于所述基因组或其部分的高灵敏拷贝数变异(cnv)分析。
263.a37.根据实施方案a36所述的方法,其中所述拷贝数变异(cnv)是扩增。
264.a38.根据实施方案a36所述的方法,其中所述拷贝数变异(cnv)是杂合或纯合缺失。
265.a39.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于所述基因组或其部分的变体发现、单倍型定相和基因组组装。
266.a39.1根据实施方案a18.1或a19所述的方法,其中所述序列信息用于包括父本基因组的第一样品和包括母本基因组的第二样品中的变体发现和单倍型定相,并且所述父本基因组和所述母本基因组的所述定相变体用于分析从母体的cfdna中获得的胎儿基因组的序列数据。
267.a40.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于所述基因组或其部分的单倍型定相和基因组组装。
268.a41.根据实施方案a18.1或a19所述的方法,其中所述序列信息用于所述基因组或其部分的基因组组装和3d基因组相互作用的检测。
269.b1.一种用于从样品制备dna分子的方法,其包括:
270.(a)使包括基因组或其部分的样品的交联dna分子与第一限制性核酸内切酶接触,从而生成交联dna分子的第一空间邻近的消化末端;
271.(b)使交联dna分子的所述第一空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点的第一交联邻位连接dna分子;
272.(c)使包括第一连接接点的所述第一交联邻位连接dna分子与第二限制性核酸内切酶接触,从而生成交联dna分子的第二空间邻近的消化末端;
273.(d)使交联dna分子的所述第二空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点和第二连接接点的第二交联邻位连接dna分子;
274.(e)使包括第一连接接点和第二连接接点的所述第二交联邻位连接dna分子与第三限制性核酸内切酶接触,从而生成交联dna分子的第三空间邻近的消化末端;
275.(f)使交联dna分子的所述第三空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点、第二连接接点和第三连接接点的第三交联邻位连接dna分子;
276.(g)使包括第一连接接点、第二连接接点和第三连接接点的所述第三交联邻位连接dna分子与第四限制性核酸内切酶接触,从而生成交联dna分子的第四空间邻近的消化末端;
277.(h)使交联dna分子的所述第四空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点、第二连接接点、第三连接接点和第四连接接点的第四交联邻位连接dna分子;
278.(i)使包括第一连接接点、第二连接接点、第三连接接点和第四连接接点的所述第四交联邻位连接dna分子与逆转交联的试剂接触,从而生成包括第一连接接点、第二连接接点、第三连接接点和第四连接接点的邻位连接dna分子;以及
279.(j)将所述邻位连接dna分子片段化,以生成邻位连接dna分子的片段,所述邻位连接dna分子的片段包括跨越所述第一连接接点、所述第二连接接点、所述第三连接接点和所述第四连接接点的片段,其中跨越所述第一连接接点、所述第二连接接点、所述第三连接接点和所述第四连接接点且长度可为用于短程测序的模板的片段包括基本上全基因组或其部分的序列。
280.b2.根据实施方案b1所述的方法,其中跨越所述第一连接接点、所述第二连接接点、所述第三连接接点和所述第四连接接点且长度为可用于短程测序的模板的所述片段包
括多达750个碱基对。
281.b3.根据实施方案b1或b2所述的方法,其中所述第一限制性核酸内切酶、所述第二限制性核酸内切酶、所述第三限制性核酸内切酶和所述第四限制性核酸内切酶选自生成具有5’突出端的末端、具有3’突出端的末端或平末端和其组合的分子的酶。
282.b4.根据实施方案b3所述的方法,其中所述第一限制性核酸内切酶、所述第二限制性核酸内切酶、所述第三限制性核酸内切酶和所述第四限制性核酸内切酶生成具有相同类型末端的分子。
283.b5.根据实施方案b3所述的方法,其中所述第一限制性核酸内切酶、所述第二限制性核酸内切酶、所述第三限制性核酸内切酶和所述第四限制性核酸内切酶中的两种或更多种生成具有不同类型末端的分子。
284.b5.1.根据实施方案b1至b5中任一项所述的方法,其中所述第一限制性核酸内切酶、所述第二限制性核酸内切酶、所述第三限制性核酸内切酶和所述第四限制性核酸内切酶中的一种或多种需要用于高活性水平的特定缓冲液,所述特定缓冲液不同于所述第一限制性核酸内切酶、所述第二限制性核酸内切酶、所述第三限制性核酸内切酶或所述第四限制性核酸内切酶中的另一种用于高活性水平所需的缓冲液。
285.b5.2.根据实施方案b1至b4中任一项所述的方法,其中所述第一限制性核酸内切酶、所述第二限制性核酸内切酶、所述第三限制性核酸内切酶和所述第四限制性核酸内切酶中的一种或多种的产物可以掺入标记,所述标记不同于所述第一限制性核酸内切酶、所述第二限制性核酸内切酶、所述第三限制性核酸内切酶或所述第四限制性核酸内切酶中的另一种所掺入的标记。
286.b6.根据实施方案b1至b5.2中任一项所述的方法,其中从选自细胞核、细胞、组织、福尔马林固定的石蜡包埋(ffpe)样品、深度福尔马林固定的样品或无细胞dna的样品中获得所述dna分子。
287.b7.根据实施方案b1至b5.4中任一项所述的方法,其中从单个细胞中获得所述dna分子。
288.b7.1.根据实施方案b1至b5.4中任一项所述的方法,其中从两个或更多个细胞中获得所述dna分子。
289.b8.根据实施方案b1至a5.4中任一项所述的方法,其中样品的所述交联dna分子包括两个或更多个基因组或其部分。
290.b9.根据实施方案b1至b8中任一项所述的方法,其中在染色质构象测定中分析所述邻位连接dna分子。
291.b10.根据实施方案b9所述的方法,其中所述染色质构象测定是capture-c、3c、4c、5c、hic、capture-hic、hichip、plac-seq、栓系染色体捕获(tcc)、hiculfite、methyl-hic、hichirp或其组合。
292.b11.根据实施方案b9所述的方法,其中所述测定是全基因组的。
293.b11.1.根据实施方案b11所述的方法,其中所述测定是3c、hic、栓系染色体捕获(tcc)、hiculfite、methyl-hic或其组合。
294.b12.根据实施方案b9所述的方法,其中所述测定针对所述基因组中的一个或多个靶区域。
295.b12.1.根据实施方案b12所述的方法,其中所述测定是capture-c、4c、5c、capture-hic、hichip、plac-seq、hichlrp或其组合。
296.b13.根据实施方案b12所述的方法,其中所述靶标是单核苷酸变异、插入、缺失、拷贝数变异、基因组重排或用于定相的靶标。
297.b14.根据实施方案b12或b13所述的方法,其中所述样品包括癌症基因组,并且所述靶区域与表型相关。
298.b15.根据实施方案b1至b14中任一项所述的方法,其中使用所述片段化的邻位连接dna分子来制备用于dna测序的模板分子文库。
299.b16.根据实施方案b15所述的方法,其中使所述片段化的邻位连接分子富集包括连接接点的片段化的邻位连接dna分子,并且使用包括连接接点的所述片段化的邻位连接dna分子来制备用于dna测序的模板分子文库。
300.b17.根据实施方案b16所述的方法,其中所述测定是hic、capture-hic、hiscip、plac-seq、hiculfite或methyl-hic,并且用亲和纯化标记物标记所述连接接点。
301.b17.1.根据实施方案b17所述的方法,其中通过用亲和纯化分子对亲和纯化标记物的亲和纯化进行富集。
302.b17.2.根据实施方案b17.1所述的方法,其中所述亲和纯化分子是链霉抗生物素蛋白。
303.b17.3.根据实施方案b16所述的方法,其中通过大小选择对包括连接接点的片段化的邻位连接dna分子进行富集。
304.b18.根据实施方案b15至b17.3中任一项所述的方法,其中所述模板分子文库提供基因组或其部分的均匀的全基因组覆盖。
305.b18.1.根据实施方案b15至a18中任一项所述的方法,其中对所述模板分子文库进行测序以生成包括序列信息的序列读段。
306.b19.根据实施方案b18.1所述的方法,其中所述测序是短读测序。
307.b20.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于所述基因组或其部分的基因组重排分析。
308.b21.根据实施方案b20所述的方法,其中所述基因组重排分析包括断点的鉴别。
309.b22.根据实施方案b21所述的方法,其中给定序列读段的序列信息位于所述断点的上游和下游。
310.b23.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于所述基因组或其部分的重叠群的聚类和排序。
311.b24.根据实施方案b23所述的方法,其中序列信息包括被聚类和排序的每个重叠群的序列信息。
312.b25.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于确定所述基因组或其部分的重叠群定向。
313.b26.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于对所述基因组或其部分的重叠群进行聚类、排序和定向。
314.b27.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于检测所述基因组或其部分的成对3d基因组相互作用。
315.b28.根据实施方案b27所述的方法,其中所述3d基因组相互作用在启动子、增强子、基因调控元件、gwas基因座、染色质环和拓扑结构域锚、重复元件、多梳区、基因体、外显子或整合的病毒序列之间。
316.b29.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于所述基因组或其部分的蛋白质因子定位分析和3d构象分析。
317.b30.根据实施方案b29所述的方法,其中所述蛋白质因子定位分析和3d构象分析包括plac-seq或hichip。
318.b31.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于所述基因组或其部分的单倍型定相。
319.b32.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于所述基因组或其部分的基因组组装和3d构象分析。
320.b33.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于所述基因组或其部分的dna甲基化分析。
321.b33.1.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于所述基因组或其部分的dna甲基化分析和3d基因组相互作用的检测。
322.b34.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于所述基因组或其部分的单核苷酸变体(snv)发现。
323.b35.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于所述基因组或其部分的远程测序信息的碱基校正。
324.b36.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于所述基因组或其部分的高灵敏拷贝数变异(cnv)分析。
325.b37.根据实施方案b36所述的方法,其中所述拷贝数变异(cnv)是扩增。
326.b38.根据实施方案b36所述的方法,其中所述拷贝数变异(cnv)是杂合或纯合缺失。
327.b39.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于所述基因组或其部分的变体发现、单倍型定相和基因组组装。
328.b40.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于所述基因组或其部分的单倍型定相和基因组组装。
329.b41.根据实施方案b18.1或b19所述的方法,其中所述序列信息用于所述基因组或其部分的基因组组装和3d基因组相互作用的检测。
330.c1.一种用于从样品制备dna分子的方法,其包括:
331.(a)使包括基因组或其部分的样品的交联dna分子与一组四种限制性核酸内切酶接触;从而生成交联dna分子的空间邻近的消化末端;
332.(b)使交联dna分子的所述空间邻近的消化末端与一种或多种试剂接触,所述一种或多种试剂将附接至核苷酸的生物素掺入到空间邻近的消化末端中,从而生成包括有标记的空间邻近的消化末端的交联dna分子;
333.(c)使包括标记的空间邻近的消化末端的所述交联dna分子与连接酶接触,从而生成包括标记的连接接点的交联邻位连接dna分子;
334.(d)使包括标记的连接接点的交联邻位连接dna分子与逆转交联的试剂接触,从而
生成包括标记的连接接点的邻位连接dna分子;
335.(e)将包括标记的连接接点的所述邻位连接dna分子片段化,以生成邻位连接dna分子的片段,所述邻位连接dna分子的片段包括跨越所述标记的连接接点的片段,其中跨越所述连接接点且长度可为用于短程测序的模板的片段包括基本上全基因组或其部分的序列;以及
336.(f)通过使用包括链霉抗生物素蛋白的亲和纯化分子对标记的连接接点进行亲和纯化,富集跨越所述标记的连接接点的dna片段。
337.c2.根据实施方案c1所述的方法,其中跨越所述连接接点的所述片段包括多达750个碱基对的片段。
338.c3.根据实施方案c1或c2所述的方法,其中所述链霉抗生物素蛋白包括链霉抗生物素蛋白包被的珠粒。
339.c4.根据实施方案c1至c3中任一项所述的方法,其中所述组的每种限制性核酸内切酶在常用缓冲液中具有高活性水平,并且所述组的每种限制性核酸内切酶具有至少1/256的理论消化频率。
340.c5.根据实施方案c1至c4中任一项所述的方法,其中所述限制性核酸内切酶是:mboi、hinfi、msei和ddei。
341.c5.1.根据实施方案c1至c4中任一项所述的方法,其中所述限制性核酸内切酶是:hpych4iv、hinfi、hinp1i和msei。
342.c6.根据实施方案c1至c5.1中任一项所述的方法,其中从选自细胞核、细胞、组织、福尔马林固定的石蜡包埋(ffpe)样品、深度福尔马林固定的样品或无细胞dna的样品中获得所述dna分子。
343.c7.根据实施方案c1至c5.1中任一项所述的方法,其中从单个细胞中获得所述dna分子。
344.c7.1.根据实施方案c1至c5.1中任一项所述的方法,其中从两个或更多个细胞中获得所述dna分子。
345.c8.根据实施方案c1至c5.1中任一项所述的方法,其中样品的所述交联dna分子包括两个或更多个基因组或其部分。
346.c9.根据实施方案c1至c8中任一项所述的方法,其中在染色质构象测定中分析所述邻位连接dna分子。
347.c10.根据实施方案c9所述的方法,其中所述染色质构象测定是capture-c、3c、4c、5c、hic、capture-hic、hichip、plac-seq、栓系染色体捕获(tcc)、hiculfite、methyl-hic、hichirp或其组合。
348.c11.根据实施方案c9所述的方法,其中所述测定是全基因组的。
349.c11.1.根据实施方案c11所述的方法,其中所述测定是3c、hic、栓系染色体捕获(tcc)、hiculfite、methyl-hic或其组合。
350.c12.根据实施方案c9所述的方法,其中所述测定针对所述基因组中的一个或多个靶区域。
351.c12.1.根据实施方案c12所述的方法,其中所述测定是capture-c、4c、5c、capture-hic、hichip、plac-seq、hichlrp或其组合。
352.c13.根据实施方案c12所述的方法,其中所述靶标是单核苷酸变异、插入、缺失、拷贝数变异、基因组重排或用于定相的靶标。
353.c14.根据实施方案c12或c13所述的方法,其中所述样品包括癌症基因组,并且所述靶区域与表型相关。
354.c15.根据实施方案c1至c14中任一项所述的方法,其中使用所述片段化的邻位连接dna分子来制备用于dna测序的模板分子文库。
355.c16.根据实施方案c15所述的方法,其中对所述片段化的邻位连接分子富集包括连接接点的片段化的邻位连接dna分子,并且使用包括连接接点的所述片段化的邻位连接dna分子来制备用于dna测序的模板分子文库。
356.c17.根据实施方案c16所述的方法,其中所述测定是hic、capture-hic、hiscip、plac-seq、hiculfite或methyl-hic,并且用亲和纯化标记物标记所述连接接点。
357.c17.1.根据实施方案c17所述的方法,其中通过用亲和纯化分子对所述亲和纯化标记物的亲和纯化进行富集。
358.c17.2.根据实施方案c17.1所述的方法,其中所述亲和纯化分子是链霉抗生物素蛋白。
359.c17.3.根据实施方案c16所述的方法,其中通过大小选择对包括连接接点的片段化的邻位连接dna分子进行富集。
360.c18.根据实施方案c15至c17.3中任一项所述的方法,其中所述模板分子文库提供基因组或其部分的均匀的全基因组覆盖。
361.c18.1.根据实施方案c15至c18中任一项所述的方法,其中对所述模板分子文库进行测序以生成包括序列信息的序列读段。
362.c19.根据实施方案c18.1所述的方法,其中所述测序是短读测序。
363.c20.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于所述基因组或其部分的基因组重排分析。
364.c21.根据实施方案c20所述的方法,其中所述基因组重排分析包括断点的鉴别。
365.c22.根据实施方案c21所述的方法,其中给定序列读段的序列信息位于所述断点的上游和下游。
366.c23.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于所述基因组或其部分的重叠群的聚类和排序。
367.c24.根据实施方案c23所述的方法,其中序列信息包括被聚类和排序的每个重叠群的序列信息。
368.c25.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于确定所述基因组或其部分的重叠群定向。
369.c26.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于对所述基因组或其部分的重叠群进行聚类、排序和定向。
370.c27.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于检测所述基因组或其部分的成对3d基因组相互作用。
371.c28.根据实施方案c27所述的方法,其中所述3d基因组相互作用在启动子、增强子、基因调控元件、gwas基因座、染色质环和拓扑结构域锚、重复元件、多梳区、基因体、外显
子或整合的病毒序列之间。
372.c29.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于所述基因组或其部分的蛋白质因子定位分析和3d构象分析。
373.c30.根据实施方案c29所述的方法,其中所述蛋白质因子定位分析和3d构象分析包括plac-seq或hichip。
374.c31.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于所述基因组或其部分的单倍型定相。
375.c32.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于所述基因组或其部分的基因组组装和3d构象分析。
376.c33.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于所述基因组或其部分的dna甲基化分析。
377.c33.1.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于所述基因组或其部分的dna甲基化分析和3d基因组相互作用的检测。
378.c34.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于所述基因组或其部分的单核苷酸变体(snv)发现。
379.c35.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于所述基因组或其部分的远程测序信息的碱基校正。
380.c36.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于所述基因组或其部分的高灵敏拷贝数变异(cnv)分析。
381.c37.根据实施方案c36所述的方法,其中所述拷贝数变异(cnv)是扩增。
382.c38.根据实施方案c36所述的方法,其中所述拷贝数变异(cnv)是杂合或纯合缺失。
383.c39.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于所述基因组或其部分的变体发现、单倍型定相和基因组组装。
384.c40.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于所述基因组或其部分的单倍型定相和基因组组装。
385.c41.根据实施方案c18.1或c19所述的方法,其中所述序列信息用于所述基因组或其部分的基因组组装和3d基因组相互作用的检测。
386.d1.一种试剂盒,其包括:
387.(a)三种或更多种限制性核酸内切酶;
388.(b)限制性核酸内切酶缓冲液;以及
389.(c)以下中的一种或多种:生物素化的核苷酸、未标记的核苷酸、dna聚合酶、连接酶、连接酶缓冲液、一种或多种附加的用于逆转交联的缓冲液和试剂。
390.d2.根据实施方案d1所述的试剂盒,其中所述限制性核酸内切酶在分开的容器中。
391.d3.根据实施方案d1所述的试剂盒,其中所述限制性核酸内切酶在单个容器中。
392.d4.根据实施方案d1至d3中任一项所述的试剂盒,其中每种限制性核酸内切酶在常用的限制性核酸内切酶缓冲液中具有高活性水平,并且每种限制性核酸内切酶具有至少1/256的理论消化频率。
393.d5.根据实施方案d1至d4中任一项所述的试剂盒,其中所述限制性核酸内切酶缓
冲液在与所述限制性核酸内切酶分开的容器中。
394.d6.根据实施方案d1至d5中任一项所述的试剂盒,其进一步包括说明书。
395.e1.一种试剂盒,其包括:
396.(a)四种限制性核酸内切酶;
397.(b)限制性核酸内切酶缓冲液;以及
398.(c)以下中的一种或多种:生物素化的核苷酸、未标记的核苷酸、dna聚合酶、连接酶、连接酶缓冲液、一种或多种附加的用于逆转交联的缓冲液和试剂。
399.e2.根据实施方案e1所述的试剂盒,其中所述四种限制性核酸内切酶在分开的容器中。
400.e3.根据实施方案e1所述的试剂盒,其中所述四种限制性核酸内切酶在单个容器中。
401.e4.根据实施方案e1至e3中任一项所述的试剂盒,其中所述限制性核酸内切酶缓冲液与所述四种限制性核酸内切酶在分开的容器中。
402.e5.根据实施方案e1至e4中任一项所述的试剂盒,其中每种限制性核酸内切酶在常用的限制性核酸内切酶缓冲液中具有高活性水平,并且每种限制性核酸内切酶具有至少1/256的理论消化频率。
403.e6.根据实施方案e1至e5中任一项所述的试剂盒,其中所述四种限制性核酸内切酶是:mboi、hinfi、msei和ddei。
404.e7.根据实施方案e1至e5中任一项所述的试剂盒,其中所述四种限制性核酸内切酶是:hpych4iv、hinfi、hinp1i和msei。
405.e8.根据实施方案e1至e7中任一项所述的试剂盒,其进一步包括说明书。
406.f1.一种试剂盒,其包括:
407.(a)四种限制性核酸内切酶;
408.(b)两种或更多种限制性核酸内切酶缓冲液;以及
409.(c)以下中的一种或多种:生物素化的核苷酸、未标记的核苷酸、dna聚合酶、连接酶、连接酶缓冲液、一种或多种附加的用于逆转交联的缓冲液和试剂。
410.f2.根据实施方案f1所述的试剂盒,其中所述四种限制性核酸内切酶在分开的容器中。
411.f3.根据实施方案f1至f3中任一项所述的试剂盒,其中所述两种或更多种限制性核酸内切酶缓冲液在与所述四种限制性核酸内切酶分开的容器中。
412.f4.根据实施方案f1至f3中任一项所述的试剂盒,其中每种限制性核酸内切酶具有至少1/256的理论消化频率。
413.f5.根据实施方案f1至f4中任一项所述的试剂盒,其中所述限制性核酸内切酶中的至少两种需要用于高水平活性的独特缓冲液。
414.f6.根据实施方案f1至f5中任一项所述的试剂盒,其进一步包括说明书。
415.g1.一种用于从样品制备dna分子的方法,其包括:
416.(a)使来自样品的具有稳定空间相互作用的空间邻近的dna分子与两种或更多种限制性核酸内切酶接触,从而消化所述dna分子,并生成dna分子的空间邻近的消化末端;以及
417.(b)使dna分子的所述空间邻近的消化末端与连接酶接触,从而生成包括连接接点的邻位连接dna分子,其中所述连接接点是未标记的。
418.g2.根据实施方案g1所述的方法,其中所述空间邻近的dna分子包括交联dna分子。
419.g2.1.根据实施方案g1或g2所述的方法,其中样品的具有稳定空间相互作用的所述空间邻近的dna分子在细胞/细胞核内,并且所述接触步骤在原位进行。
420.g2.2.根据实施方案g1或g2所述的方法,其中所述空间邻近的dna分子包括基因组或其部分。
421.g3.根据实施方案g1至g2.2中任一项所述的方法,其中存在两种限制性核酸内切酶。
422.g4.根据实施方案g1至g2.2中任一项所述的方法,其中存在至少三种限制性核酸内切酶。
423.g4.1.根据实施方案g4所述的方法,其中存在三种限制性核酸内切酶。
424.g5.根据实施方案g1至g4.1中任一项所述的方法,其中所述限制性核酸内切酶中的一种是nlaiii。
425.g6.根据实施方案g1至g5中任一项所述的方法,其中所述限制性核酸内切酶中的一种是nlaiii,并且另一种限制性核酸内切酶是mboi或msei。
426.g7.根据实施方案g1至g4.1中任一项所述的方法,其中所述限制性核酸内切酶中的一种是nlaiii,并且另一种限制性核酸内切酶是mboi或msei。
427.g8.根据实施方案g4或g4.1所述的方法,其中所述限制性核酸内切酶是:nlaiii、mboi和msei。
428.g9.根据实施方案g1至g5中任一项所述的方法,其中所述限制性核酸内切酶产生相同的突出序列。
429.g10.根据实施方案g1至g8中任一项所述的方法,其中所述限制性核酸内切酶产生不同的突出序列。
430.g11.根据实施方案g1至g10中任一项所述的方法,其中与所有所述限制性核酸内切酶的接触和消化是同时的。
431.g12.根据实施方案g1至g10中任一项所述的方法,其中与各种限制性核酸内切酶的接触和消化是按顺序的。
432.g12.1.根据实施方案g12所述的方法,其中用先前的一种或多种核酸内切酶进行的所述消化已基本上完成。
433.g12.2.根据实施方案g12所述的方法,其中用先前的一种或多种核酸内切酶进行的所述消化还未完成。
434.g13.根据实施方案g4至g10中任一项所述的方法,其中与限制性核酸内切酶的接触和消化是按顺序的,并且至少一次接触和消化是用至少两种限制性核酸内切酶进行。
435.g14.根据实施方案g12至g13中任一项所述的方法,其中所述按顺序的接触和消化对于所述限制性核酸内切酶来说具有确定的顺序。
436.g14.1.根据实施方案g11所述的方法,其中与连接酶的接触在完成通过限制性核酸内切酶进行的消化之后。
437.g14.2.根据实施方案g12至g14中任一项所述的方法,其中与连接酶的接触在完成
与所有所述限制性核酸内切酶的所述按顺序的接触和消化之后。
438.g15.根据实施方案g12至g14中任一项所述的方法,其中在每次与一种或多种限制性核酸内切酶接触和消化之后,与连接酶接触。
439.g16.根据实施方案g1至g15中任一项所述的方法,其中从选自细胞核、细胞、组织、福尔马林固定的石蜡包埋(ffpe)样品、深度福尔马林固定的样品或无细胞dna的样品中获得所述dna分子。
440.g16.1根据实施方案g16所述的方法,其中所述样品在水溶液中或附着到固体表面。
441.g17.根据实施方案g1至g16.1中任一项所述的方法,其中从单个细胞中获得所述dna分子。
442.g18.根据实施方案g1至g16.1中任一项所述的方法,其中从两个或更多个细胞中获得所述dna分子。
443.g19.根据实施方案g1至g18中任一项所述的方法,其中样品的所述dna分子包括两个或更多个基因组或其部分。
444.g20.根据实施方案g1至g19中任一项所述的方法,其中所述方法包括一个或多个针对4c、5c、capture-c、3c-chip或methyl-3c方法的步骤。
445.g21.根据实施方案g1至g20中任一项所述的方法,其中包括连接接点的所述邻位连接dna分子来源于基本上代表整个基因组的序列。
446.g22.根据实施方案g1至g21中任一项所述的方法,其中纯化所述包括连接接点的邻位连接dna分子。
447.g23.根据实施方案g2至g22中任一项所述的方法,其中使包括连接接点的所述交联的邻位连接dna分子与逆转交联的试剂接触。
448.g24.根据实施方案g1至g23中任一项所述的方法,其中使包括连接接点的邻位连接dna分子富集具有连接接点的dna分子。
449.g24.1.根据实施方案g24所述的方法,其中通过大小选择来富集具有连接接点的dna分子。
450.g24.2.根据实施方案g24.1所述的方法,其中大小选择包括使用珠粒。
451.g24.3.根据实施方案g24.1所述的方法,其中大小选择包括凝胶提取或大小选择性dna沉淀。
452.g25.根据实施方案g1至g24.3中任一项所述的方法,其中由所述邻位连接dna分子制备用于dna测序的模板分子文库。
453.g25.1.根据实施方案g25所述的方法,其中在构建所述文库时在扩增步骤之前或之后进行大小选择以富集具有连接接点的dna分子。
454.g26.根据实施方案g25或g25.1所述的方法,其中对所述模板分子文库进行测序以生成包括序列信息的序列读段。
455.g27.根据实施方案g26所述的方法,其中所述测序是短读测序。
456.g27.1.根据实施方案g1至g27中任一项所述的方法,其中至少30%的所述核酸模板是远程顺式分子。
457.g27.2.根据实施方案g1至g27中任一项所述的方法,其中至少40%的所述核酸模
板是远程顺式分子。
458.g27.3.根据实施方案g1至g27中任一项所述的方法,其中至少50%的所述核酸模板是远程顺式分子。
459.g27.4.根据实施方案g1至g27中任一项所述的方法,其中至少60%的所述核酸模板是远程顺式分子。
460.g27.5.根据实施方案g27所述的方法,其中在制备文库之前,将所述邻位连接dna分子片段化以生成邻位连接dna分子的片段,所述邻位连接dna分子的片段包括跨越所述连接接点的片段。
461.g27.6.根据实施方案g26所述的方法,其中所述测序是长读测序。
462.g28.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于检测所述基因组或其部分的成对3d基因组相互作用。
463.g29.根据实施方案g28所述的方法,其中所述3d基因组相互作用在启动子、增强子、基因调控元件、gwas基因座、染色质环和拓扑结构域锚、重复元件、多梳区、基因体、外显子或整合的病毒序列之间。
464.g30.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于所述基因组或其部分的蛋白质因子定位分析和3d构象分析。
465.g31.根据实施方案g30所述的方法,其中所述蛋白质因子定位分析和3d构象分析包括3c-chip。
466.g32.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于所述基因组或其部分的基因组重排分析。
467.g33.根据实施方案g32所述的方法,其中所述基因组重排分析包括断点的鉴别。
468.g34.根据实施方案g33所述的方法,其中给定序列读段的序列信息位于所述断点的上游和下游。
469.g35.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于所述基因组或其部分的重叠群的聚类和排序。
470.g36.根据实施方案g35所述的方法,其中序列信息包括被聚类和排序的每个重叠群的序列信息。
471.g37.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于确定所述基因组或其部分的重叠群定向。
472.g38.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于对所述基因组或其部分的重叠群进行聚类、排序和定向。
473.g39.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于所述基因组或其部分的单倍型定相。
474.g40.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于所述基因组或其部分的dna甲基化分析。
475.g41.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于所述基因组或其部分的单核苷酸变体(snv)发现。
476.g42.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于所述基因组或其部分的远程测序信息的碱基校正。
477.g43.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于所述基因组或其部分的高灵敏拷贝数变异(cnv)分析。
478.g44.根据实施方案g43所述的方法,其中所述拷贝数变异(cnv)是扩增。
479.g45.根据实施方案g43所述的方法,其中所述拷贝数变异(cnv)是杂合或纯合缺失。
480.g46.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于所述基因组或其部分的变体发现、单倍型定相和基因组组装。
481.g47.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于包括父本基因组的第一样品和包括母本基因组的第二样品中的变体发现和单倍型定相,并且所述父本基因组和所述母本基因组的所述定相变体用于分析从母体的cfdna中获得的胎儿基因组的序列数据。
482.g48.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于所述基因组或其部分的单倍型定相和基因组组装。
483.g49.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于所述基因组或其部分的基因组组装和3d构象分析。
484.g50.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于所述基因组或其部分的dna甲基化分析和3d基因组相互作用的检测。
485.g51.根据实施方案g26至g27.6中任一项所述的方法,其中所述序列信息用于基因组或其部分的基因组组装和3d基因组相互作用的检测。
486.g52.根据实施方案g1至g51中任一项所述的方法,其中邻位连接dna分子的分子邻接性被保持在条形码中。
487.g53.根据实施方案g52所述的方法,其中通过在文库制备之前使邻位连接dna与条形码化转座体连接珠粒接触,将条形码引入到所述邻位连接dna分子中。
488.g54.根据实施方案g52至g53所述的方法,其中通过利用所保持的邻位连接dna分子的分子邻接性,将所述序列信息用于检测基因组或其部分的更高级3d基因组相互作用。
489.g55.根据实施方案g52至g54中任一项所述的方法,其中通过利用所保持的邻位连接dna分子的分子邻接性,将所述序列信息用于检测所述基因组或其部分的三种或更多种同时进行的3d基因组相互作用。
490.g56.根据实施方案g52至g55中任一项所述的方法,其中通过利用所保持的邻位连接dna分子的分子邻接性,将所述序列信息用于检测虚拟的成对3d基因组相互作用。
491.g57.根据实施方案g56所述的方法,其中虚拟的成对3d基因组相互作用在所述基因组或其部分的给定邻位连接dna分子内彼此不直接连接的限制性片段之间。
492.g58.根据实施方案g52至g57中任一项所述的方法,其中通过利用所保持的邻位连接dna分子的分子邻接性获得的所述成对相互作用、虚拟的成对相互作用、和/或更高级相互作用用于所述基因组或其部分的3d基因组相互作用、所述基因组或其部分的基因组重排分析、所述基因组或其部分的重叠群的聚类和排序、确定所述基因组或其部分的重叠群定向、所述基因组或其部分的单倍型定相、所述基因组或其部分的dna甲基化分析、所述基因组或其部分的单核苷酸变体(snv)发现、所述基因组或其部分的远程测序信息的碱基校正、所述基因组或其部分的高灵敏拷贝数变异(cnv)分析或其组合。
493.h1.一种用于从样品制备dna分子的方法,其包括:
494.(a)使来自样品的具有稳定空间相互作用的空间邻近的dna分子与第一限制性核酸内切酶接触,从而消化所述dna分子,并生成dna分子的第一空间邻近的消化末端;
495.(b)使dna分子的所述第一空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点的第一邻位连接dna分子,其中所述连接接点是未标记的;
496.(c)使包括第一连接接点的所述第一邻位连接dna分子与第二限制性核酸内切酶接触,从而消化所述第一邻位连接dna分子,并生成dna分子的第二空间邻近的消化末端;以及
497.(d)使dna分子的所述第二空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点和第二连接接点的第二邻位连接dna分子,其中所述连接接点是未标记的。
498.h2.根据实施方案h1所述的方法,其包括:
499.(e)使包括第一连接接点和第二连接接点的所述第二邻位连接dna分子与第三限制性核酸内切酶接触,从而消化所述第二邻位连接dna分子,并生成dna分子的第三空间邻近的消化末端;以及
500.(f)使dna分子的所述第三空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点、第二连接接点和第三连接接点的第三邻位连接dna分子,其中所述连接接点是未标记的。
501.h3.一种用于从样品制备dna分子的方法,其包括:
502.(a)使在来自样品的细胞/细胞核内具有稳定空间相互作用的空间邻近的dna分子与第一限制性核酸内切酶接触,从而消化所述dna分子,并生成dna分子的第一空间邻近的消化末端;
503.(b)使dna分子的所述第一空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点的第一邻位连接dna分子,其中所述连接接点是未标记的,并且所述接触步骤在原位进行;
504.(c)使包括第一连接接点的所述第一邻位连接dna分子与第二限制性核酸内切酶接触,从而消化所述第一邻位连接dna分子,并生成dna分子的第二空间邻近的消化末端;以及
505.(d)使dna分子的所述第二空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点和第二连接接点的第二邻位连接dna分子,其中所述连接接点是未标记的,并且所述接触步骤在原位进行。
506.h4.根据实施方案h3所述的方法,其包括:
507.(e)使包括第一连接接点和第二连接接点的所述第二邻位连接dna分子与第三限制性核酸内切酶接触,从而消化所述第二邻位连接dna分子,并生成dna分子的第三空间邻近的消化末端;以及
508.(f)使dna分子的所述第三空间邻近的消化末端与连接酶接触,从而生成包括第一连接接点、第二连接接点和第三连接接点的第三邻位连接dna分子,其中所述连接接点是未标记的,并且所述接触步骤在原位进行。
509.h5.根据实施方案h1至h4中任一项所述的方法,其中所述限制性核酸内切酶产生相同的突出序列。
510.h6.根据实施方案h1至h4中任一项所述的方法,其中所述限制性核酸内切酶产生不同的突出序列。
511.i1.一种获得从邻位连接组织切片得到的序列信息的空间定位的方法,其包括:
512.(a)使固体支持物上的组织切片与两种或更多种限制性核酸内切酶接触,所述组织切片包括具有空间邻近的dna分子的细胞/细胞核,所述空间邻近的dna分子具有稳定的空间相互作用,从而消化所述dna分子,并生成具有dna分子的空间邻近的消化末端的组织切片;
513.(b)使所述组织切片的dna分子的所述空间邻近的消化末端与连接酶接触,从而生成具有包括连接接点的邻位连接dna分子的组织切片,其中所述连接接点是未标记的或标记的,并且所述接触步骤在原位进行。
514.(c)将所述组织切片显微解剖成空间上不同的区域;
515.(d)从一个或多个空间上不同的区域获得邻位连接dna分子;
516.(e)对使用所述邻位连接dna分子制备的文库进行测序,以生成
517.序列信息;以及
518.(f)将来自邻位连接分子的所述序列信息分配给从中获得所述邻位连接分子的所述组织样品的空间上不同的区域,从而获得序列信息的所述空间定位。
519.i2.一种获得从邻位连接组织切片得到的序列信息的空间定位的方法,其包括:
520.(a)使固体支持物上的组织切片与两种或更多种限制性核酸内切酶接触,所述组织切片包括具有空间邻近的dna分子的细胞/细胞核,所述空间邻近的dna分子具有稳定的空间相互作用,从而消化所述dna分子,并生成具有dna分子的空间邻近的消化末端的组织切片;
521.(b)使所述组织切片的dna分子的所述空间邻近的消化末端与连接酶接触,从而生成具有包括连接接点的邻位连接dna分子的组织切片,其中所述连接接点是未标记的或标记的,并且所述接触步骤在原位进行。
522.(c)将所述组织切片显微解剖成空间上不同的区域;
523.(d)从空间上不同的区域获得包括邻位连接dna分子的单个细胞;
524.(e)对使用单个细胞的所述邻位连接dna分子制备的文库进行测序,以生成来自单个细胞的序列信息;以及
525.(f)将来自单个细胞的所述序列信息分配给从中获得所述细胞的所述组织样品的空间上不同的区域,从而获得来自单个细胞的序列信息的所述空间定位。
526.i3.一种获得从邻位连接组织切片得到的序列信息的空间定位的方法,其包括:
527.(a)将组织切片显微解剖成空间上不同的区域,所述组织切片包括具有空间邻近的dna分子的细胞/细胞核,所述空间邻近的dna分子具有稳定的空间相互作用;
528.(b)使包括具有空间邻近的dna分子的细胞/细胞核的空间上不同的区域与两种或更多种限制性核酸内切酶接触,从而消化所述dna分子,并生成具有dna分子的空间邻近的消化末端的空间上不同的区域;
529.(c)使所述空间上不同的区域的dna分子的所述空间邻近的消化末端与连接酶接触,从而生成具有包括连接接点的邻位连接dna分子的空间上不同的区域,其中所述连接接点是未标记的或标记的,并且所述接触步骤在原位进行;
530.(d)从一个或多个空间上不同的区域获得邻位连接dna分子;
531.(e)对使用来自空间上不同的区域的所述邻位连接dna分子制备的文库进行测序,以生成序列信息;以及
532.(f)将来自邻位连接分子的所述序列信息分配给从中获得所述邻位连接分子的所述组织样品的空间上不同的区域,从而获得序列信息的所述空间定位。
533.i4.一种获得从邻位连接组织切片得到的序列信息的空间定位的方法,其包括:
534.(a)将组织切片显微解剖成空间上不同的区域,所述组织切片包括具有空间邻近的dna分子的细胞/细胞核,所述空间邻近的dna分子具有稳定的空间相互作用;
535.(b)使包括具有空间邻近的dna分子的细胞/细胞核的空间上不同的区域与两种或更多种限制性核酸内切酶接触,从而消化所述dna分子,并生成具有dna分子的空间邻近的消化末端的空间上不同的区域;
536.(c)使所述空间上不同的区域的dna分子的所述空间邻近的消化末端与连接酶接触,从而生成具有包括连接接点的邻位连接dna分子的空间上不同的区域,其中所述连接接点是未标记的或标记的,并且所述接触步骤在原位进行;
537.(d)从空间上不同的区域获得包括邻位连接dna分子的单个细胞;
538.(e)对使用单个细胞的所述邻位连接dna分子制备的文库进行测序,以生成来自单个细胞的序列信息;以及
539.(f)将来自单个细胞的所述序列信息分配给从中获得所述细胞的所述组织样品的空间上不同的区域,从而获得来自单个细胞的序列信息的所述空间定位。
540.j1.一种用于测序的dna模板分子文库,其通过包括根据实施方案a1至a18中任一方法所述的方法制备。
541.j2.一种用于测序的dna模板分子文库,其通过包括根据实施方案b1至b14中任一方法所述的方法制备。
542.j3.一种用于测序的dna模板分子文库,其通过包括根据实施方案c1至c14中任一方法所述的方法制备。
543.j4.一种用于测序的dna模板分子文库,其通过包括根据实施方案g1至g27.5中任一方法所述的方法制备。
544.j5.一种用于测序的dna模板分子文库,其通过包括根据实施方案h1至h16任一方法所述的方法制备。
545.k1.一种试剂盒,其包括以下中的一种或多种:
546.(a)两种或更多种限制性核酸内切酶;
547.(b)限制性核酸内切酶缓冲液;以及
548.(c)以下中的一种或多种:未标记的核苷酸、dna聚合酶、连接酶、一种或多种附加的用于逆转交联的缓冲液和试剂、tn5转座子、具有条形码寡核苷酸的引物,其中所述试剂盒不包括生物素化的核苷酸或标记的核苷酸。
549.k2.一种试剂盒,其包括以下中的一种或多种:
550.(a)两种限制性核酸内切酶;
551.(b)限制性核酸内切酶缓冲液;以及
552.(c)以下中的一种或多种:未标记的核苷酸、dna聚合酶、连接酶、一种或多种附加
的用于逆转交联的缓冲液和试剂、tn5转座子、具有条形码寡核苷酸的引物,其中所述试剂盒不包括生物素化的核苷酸或标记的核苷酸。
553.k2.1.根据实施方案k2所述的试剂盒,其中所述限制性核酸内切酶中的一种是nlaiii。
554.k2.2.根据实施方案k2.1所述的试剂盒,其中另一种限制性核酸内切酶是mboi或msei。
555.k3.一种试剂盒,其包括以下中的一种或多种:
556.(a)三种限制性核酸内切酶;
557.(b)限制性核酸内切酶缓冲液;以及
558.(c)以下中的一种或多种:未标记的核苷酸、dna聚合酶、连接酶、一种或多种附加的用于逆转交联的缓冲液和试剂、tn5转座子、具有条形码寡核苷酸的引物,其中所述试剂盒不包括生物素化的核苷酸或标记的核苷酸。
559.k3.1.根据实施方案k3所述的试剂盒,其中所述限制性核酸内切酶中的一种是nlaiii。
560.k3.2.根据实施方案k3.1所述的试剂盒,其中所述核酸内切酶中的一种是mboi或msei。
561.k3.3.根据实施方案k3所述的试剂盒,其中所述限制性核酸内切酶是:nlaiii、mboi和msei。
562.k4.根据实施方案k1至k3.3中任一项所述的试剂盒,其中所述试剂盒的所述限制性核酸内切酶产生相同的突出序列。
563.k5.根据实施方案k1至k3.3中任一项所述的试剂盒,其中所述试剂盒的所述限制性核酸内切酶产生不同的突出序列。
564.k6.根据实施方案k1至k5中任一项所述的试剂盒,其中可以同时用所述试剂盒的所述两种或更多种限制性核酸内切酶实施消化。
565.k7.根据实施方案k1至k5中任一项所述的试剂盒,其中不能同时用所述试剂盒的一种或多种限制性核酸内切酶实施消化。
566.k8.根据实施方案k1至k7中任一项所述的试剂盒,其中所述试剂盒的所述限制性核酸内切酶在分开的容器中。
567.k9.根据实施方案k6所述的试剂盒,其中所述试剂盒的所述限制性核酸内切酶在单个容器中。
568.k10.根据实施方案k1至k7中任一项所述的试剂盒,其中所述试剂盒的所述限制性核酸内切酶在超过一个容器中。
569.k10.1.根据实施方案k10所述的试剂盒,其中至少一个容器装有超过一种限制性核酸内切酶。
570.k11.根据实施方案k1至k6中任一项所述的试剂盒,其中所述试剂盒的每种限制性核酸内切酶在常用的限制性核酸内切酶缓冲液中具有高活性水平,并且所述缓冲液在一个容器中。
571.k12.根据实施方案k1至k10.1中任一项所述的试剂盒,其中超过一种限制性核酸内切酶缓冲液在所述试剂盒中,并且所述缓冲液在分开的容器中。
572.k13.根据实施方案k1至k12中任一项所述的试剂盒,其中限制性核酸内切酶缓冲液与限制性核酸内切酶在分开的容器中。
573.k14.根据实施方案k1至k13中任一项所述的试剂盒,其中所述试剂盒包括说明书。
574.k14.1.根据实施方案k14所述的试剂盒,其中所述说明书描述了试剂盒的所述限制性酶待使用的顺序。
575.***
576.本文引用的每个专利、专利申请、出版物和文献的全部内容在此以引用方式并入。以上专利、专利申请、出版物和文件的引用不是承认任何前述内容是相关的现有技术,它也不构成对这些出版物或文献的内容或日期的任何承认。他们的引用并非指示搜索相关公开。所有关于文件的日期或内容的声明都是基于可获得的信息,而并非对它们的准确性或正确性的承认。
577.可在不脱离所述技术的基本方面的情况下对上文进行修改。尽管已参考一个或多个具体实施方案详细描述了所述技术,但本领域普通技术人员将认识到可对本技术中具体公开的实施方案作出改变,但这些修改和改进在所述技术的范围和精神内。
578.本文说明性地描述的技术适当地可在不存在本文未具体公开的任何要素的情况下实施。因此,例如,在本文中的每种情况下,术语“包括”、“基本上由...组成”和“由...组成”中的任一个均可用其他两个术语中的任一个替代。已采用的术语和表达被用作描述而非限制的术语,并且使用此类术语和表达并不排除所显示和描述的特征或其部分的任何等同物,并且在所要求保护的技术的范围内进行各种修改是可能的。除非上下文中明显描述一个所述要素或超过一个所述要素,否则术语“一(a或an)”可指其修饰的一个或多个要素(例如,“试剂”可意指一种或多种试剂)。如本文所用的术语“约”是指基础参数的10%内(即,加或减10%)的值,并且在值串的开头使用术语“约”修饰每个值(即,“约1、2和3”是指约1、约2和约3)。例如,“约100克”的重量可包括90克到110克的重量。此外,当本文描述值的列表(例如,约50%、60%、70%、80%、85%或86%)时,该列表包括其所有中间值和分数值(例如,54%、85.4%)。因此,应当理解,尽管已通过代表性实施方案和任选特征具体地公开了本技术,但本领域技术人员可采用本文所公开的构思的修改和变更,并且认为此类修改和变更在本技术的范围内。
579.本技术的某些实施方案陈述在所附的权利要求中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1