一种酸性莼菜多糖及其分离纯化方法和应用
1.本发明涉及多糖分离纯化方法及应用领域,特别涉及一种酸性莼菜多糖及其分离纯化方法和应用。
背景技术:
2.多糖是植物细胞壁和细胞中普遍存在的重要成分,其理化特性和生物活性因其组成结构的不同而有所不同。研究表明,多糖具有抗肿瘤、抗氧化和免疫调节作用等多种生物活性。近年来,新型多糖的提取、纯化、结构分析和药物研究越来越受到关注。
3.莼菜是多年生水生宿根草本,主要分布于中国江西、浙江、江苏等地。它生长在池塘、湖泊和沼泽中,是一种珍贵的食品和药物两用植物,具有清热解毒、利水、消肿等功效。莼菜富含多种化学成分,如多糖、多酚、蛋白质、氨基酸、维生素和微量元素,多糖作为其主要的营养成分,被认为具有多种保健和药用价值。近年来,大量研究表明莼菜多糖具有抗氧化、抗肿瘤、降血压、降血糖等生物活性,莼菜多糖在保健与医疗方面的应用具有广阔的研究前景。
4.然而,尽管之前对于莼菜多糖的提取和分离有了一定的研究,但对于莼菜嫩叶中酸性多糖的结构及生物活性研究尚未见相关报道,特别是其体外抗氧化和抗肿瘤活性方面的研究,并且对于其结构与生物活性之间的相关性还没有明确的解释。这些问题都阻碍了莼菜资源的综合利用与深加工。
5.中国专利申请号201910344565.4,专利名称一种莼菜多糖的提取方法,公开了如下提取步骤:莼菜先用氢氧化钠溶液洗涤,以酶提法提取,经脱蛋白、超滤膜超滤、醇沉等步骤得到高纯度的莼菜粗多糖。
6.尽管上述发明可以得到纯度较高的莼菜粗多糖,但对莼菜粗多糖没有进行进一步的分离纯化,对多糖的结构没有深入的研究,活性成分不明确。
技术实现要素:
7.为了解决现有莼菜多糖分离纯化方法,其多糖成分未得到有效应用的技术问题,本发明提供了一种用于莼菜多糖分离纯化的方法,得到一种新型酸性莼菜多糖,同时研究其体外抗氧化和抑制肿瘤细胞增殖的生物活性。
8.酸性莼菜多糖的分离纯化方法,按以下具体步骤进行:
9.一、制备莼菜粗多糖
10.①
将新鲜莼菜嫩叶采用自来水洗净,沥干水分,无水乙醇回流脱脂,烘干,粉碎机粉碎,过100目筛,即得莼菜干粉;
11.②
向莼菜干粉中加入蒸馏水,料液比为1:50,加入莼菜干粉质量1~3%的果胶酶,调节溶液ph值3.5~5.5,控制提取温度45~65℃,提取1~3h,90℃灭活10min,过滤去除莼菜干粉,滤液减压浓缩至原体积1/5;
12.③
向浓缩液中边搅拌边加入三倍体积的无水乙醇,4℃静置过夜,离心,得褐色沉
淀物;
13.④
沉淀物以适量蒸馏水充分溶解,冷冻干燥,即得莼菜粗多糖;
14.二、将莼菜粗多糖以20~30倍蒸馏水复溶,然后向溶液中加入savage试剂,充分搅拌1h,离心,取上清液,冷冻干燥,即得脱蛋白后的莼菜粗多糖;
15.莼菜粗多糖溶液与savage试剂的体积比为5:1,savage试剂中氯仿与正丁醇体积比为5:1。
16.步骤二的操作重复5~10次,直至离心后液面中间无白色固体。
17.三、将步骤二获得的莼菜粗多糖以蒸馏水完全溶解,离心后将上清液一次性上样于deae-52纤维柱中,控制流速为1ml/min,分别使用蒸馏水及浓度为0.1、0.2、0.3、0.4、0.5mol/l的nacl溶液梯度洗脱,每10min收集一管,采用苯酚-硫酸法隔管检测洗脱液中多糖含量,绘制洗脱曲线,合并曲线中呈单一峰的洗脱组分,减压浓缩后蒸馏水透析72h,冷冻干燥,即得新型酸性莼菜多糖,完成所述一种新型酸性莼菜多糖的分离纯化方法。
18.单糖分析显示,该种多糖主要由d-甘露糖、d-半乳糖、d-木糖和l-岩藻糖组成,其摩尔比为0.08:1.39:1:0.23。红外分析结果表明,该种多糖为α构型的吡喃糖。
19.上述方法制备的新型酸性莼菜多糖作为天然抗氧化剂及抑制肿瘤细胞增殖的生物活性成分应用于食品、药品、保健品等领域。
20.本发明的有益效果是:
21.本发明建立了一种莼菜多糖的分离纯化方法,使用酶提法提取粗多糖,经脱蛋白,deae-52纤维素柱,透析,冻干等操作后,得到了一种纯度极高的新型酸性莼菜多糖,经分析该种多糖分子量约119.5kda,总糖含量为93.25%并含有30.91%的糖醛酸,研究表明含有糖醛酸结构单元的低聚糖或多糖常显示出十分重要的生物活性,如抗氧化、抗肿瘤、免疫调节等,因此该多糖展现出潜在的抗氧化及抗肿瘤应用价值。
22.经验证,该种多糖具有抗氧化活性,对dpph、
·
oh和abts
+
自由基具有显著的清除作用,且清除率具有浓度依赖性,其半数抑制浓度(ic
50
)分别为1.68、2.28和2.08mg/ml,该多糖对fe
2+
也具有显著的螯合能力,螯合率随多糖浓度增加而增加,其半数螯合浓度为1.80mg/ml。同时该种莼菜酸性多糖对hepg2、hela和22rv1肿瘤细胞显示出增殖抑制活性,表现出一定的剂量依赖效应,并且随着给药时间的增加,细胞存活率出现不同程度的下降。综合以上结果说明,该种新型酸性莼菜多糖作为一种全新的多糖,表现出显著的体外抗氧化活性,同时肿瘤细胞增殖抑制研究表明该种多糖可以作为一种广谱的新型抗肿瘤药物。因此,该种新型多糖可以应用于食品、药品、保健品等领域,同时还能推进莼菜深加工,提高莼菜资源的综合利用。
附图说明:
23.图1为莼菜粗多糖deae-52纤维素柱洗脱曲线;
24.图2为实例10得到的精制酸性莼菜多糖bsp-5的紫外全波长扫描图;
25.图3为实例10得到的精制酸性莼菜多糖bsp-5的gpc谱图;
26.图4为实例10得到的精制酸性莼菜多糖bsp-5及各单糖pmp衍生化后的hplc谱图;
27.图5为实例10得到的精制酸性莼菜多糖bsp-5红外谱图;
28.图6为bsp-5对dpph自由基抑制活性实验结果图;
29.图7为bsp-5对羟基自由基抑制活性实验结果图;
30.图8为bsp-5对abts
+
自由基抑制活性实验结果图;
31.图9为bsp-5对fe
2+
螯合能力实验结果图;
32.图10为bsp-5对hepg2肿瘤细胞增殖抑制实验结果图;
33.图11为bsp-5对hela肿瘤细胞增殖抑制实验结果图;
34.图12为bsp-5对22rv1肿瘤细胞增殖抑制实验结果图。
具体实施方式
35.下面将结合实施例对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。
36.实施例1:莼菜粗多糖的提取
37.1、将新鲜莼菜嫩叶采用自来水洗净,沥干水分,无水乙醇回流脱脂,烘干,粉碎机粉碎,过100目筛,即得莼菜干粉;
38.2、称取30g莼菜干粉,向莼菜干粉中加入蒸馏水,料液比为1:50,调节ph值为4.5,加入底物质量3%的果胶酶,控制提取温度45℃,提取3h,90℃灭活10min,过滤去除莼菜干粉,滤液减压浓缩至原体积1/5;
39.3、向浓缩液中边搅拌边加入三倍体积的无水乙醇,4℃静置过夜,离心,得褐色沉淀物;
40.4、沉淀物以20ml蒸馏水充分溶解,冷冻干燥,即得莼菜粗多糖;
41.5、将莼菜粗多糖充分溶解于50ml蒸馏水中,加入savage试剂10ml,其中,savage试剂中氯仿与正丁醇体积比为5:1,室温充分搅拌1h,离心,取上清液,重复10次,直至离心后液面中间无白色固体,上清液冷冻干燥,即得脱蛋白后的莼菜粗多糖3.8g,提取率12.7%;
42.实施例2:莼菜粗多糖的提取
43.1、称取实施例1所制得的莼菜干粉20g,向莼菜干粉中加入蒸馏水,料液比为1:50,调节ph值为5.5,加入底物质量2%的果胶酶,控制提取温度45℃,提取2h,90℃灭活10min,过滤去除莼菜干粉,滤液减压浓缩至原体积1/5;
44.3、向浓缩液中边搅拌边加入三倍体积的无水乙醇,4℃静置过夜,离心,得褐色沉淀物;
45.4、沉淀物以15ml蒸馏水充分溶解,冷冻干燥,即得莼菜粗多糖;
46.5、将莼菜粗多糖充分溶解于40ml蒸馏水中,加入savage试剂8ml,其中,savage试剂中氯仿与正丁醇体积比为5:1,室温充分搅拌1h,离心,取上清液,重复7次,直至离心后液面中间无白色固体,上清液冷冻干燥,即得脱蛋白后的莼菜粗多糖2.3g,提取率11.5%;
47.实施例3:莼菜粗多糖的提取
48.1、称取实施例1所制得的莼菜干粉25g,向莼菜干粉中加入蒸馏水,料液比为1:50,调节ph值为3.5,加入底物质量1%的果胶酶,控制提取温度45℃,提取1h,90℃灭活10min,过滤去除莼菜干粉,滤液减压浓缩至原体积1/5;
49.3、向浓缩液中边搅拌边加入三倍体积的无水乙醇,4℃静置过夜,离心,得褐色沉淀物;
50.4、沉淀物以10ml蒸馏水充分溶解,冷冻干燥,即得莼菜粗多糖;
51.5、将莼菜粗多糖充分溶解于45ml蒸馏水中,加入savage试剂9ml,其中,savage试剂中氯仿与正丁醇体积比为5:1,室温充分搅拌1h,离心,取上清液,重复8次,直至离心后液面中间无白色固体,上清液冷冻干燥,即得脱蛋白后的莼菜粗多糖2.1g,提取率8.4%;
52.实施例4:莼菜粗多糖的提取
53.1、称取实施例1所制得的莼菜干粉40g,向莼菜干粉中加入蒸馏水,料液比为1:50,调节ph值为5.5,加入底物质量1%的果胶酶,控制提取温度65℃,提取3h,90℃灭活10min,过滤去除莼菜干粉,滤液减压浓缩至原体积1/5;
54.3、向浓缩液中边搅拌边加入三倍体积的无水乙醇,4℃静置过夜,离心,得褐色沉淀物;
55.4、沉淀物以15ml蒸馏水充分溶解,冷冻干燥,即得莼菜粗多糖;
56.5、将莼菜粗多糖充分溶解于40ml蒸馏水中,加入savage试剂8ml,其中,savage试剂中氯仿与正丁醇体积比为5:1,室温充分搅拌1h,离心,取上清液,重复7次,直至离心后液面中间无白色固体,上清液冷冻干燥,即得脱蛋白后的莼菜粗多糖2.5g,提取率6.3%;
57.实施例5:莼菜粗多糖的提取
58.1、称取实施例1所制得的莼菜干粉25g,向莼菜干粉中加入蒸馏水,料液比为1:50,调节ph值为3.5,加入底物质量3%的果胶酶,控制提取温度65℃,提取2h,90℃灭活10min,过滤去除莼菜干粉,滤液减压浓缩至原体积1/5;
59.3、向浓缩液中边搅拌边加入三倍体积的无水乙醇,4℃静置过夜,离心,得褐色沉淀物;
60.4、沉淀物以10ml蒸馏水充分溶解,冷冻干燥,即得莼菜粗多糖;
61.5、将莼菜粗多糖充分溶解于55ml蒸馏水中,加入savage试剂11ml,其中,savage试剂中氯仿与正丁醇体积比为5:1,室温充分搅拌1h,离心,取上清液,重复6次,直至离心后液面中间无白色固体,上清液冷冻干燥,即得脱蛋白后的莼菜粗多糖2.9g,提取率11.6%;
62.实施例6:莼菜粗多糖的提取
63.1、称取实施例1所制得的莼菜干粉30g,向莼菜干粉中加入蒸馏水,料液比为1:50,调节ph值为5.5,加入底物质量3%的果胶酶,控制提取温度55℃,提取1h,90℃灭活10min,过滤去除莼菜干粉,滤液减压浓缩至原体积1/5;
64.3、向浓缩液中边搅拌边加入三倍体积的无水乙醇,4℃静置过夜,离心,得褐色沉淀物;
65.4、沉淀物以10ml蒸馏水充分溶解,冷冻干燥,即得莼菜粗多糖;
66.5、将莼菜粗多糖充分溶解于50ml蒸馏水中,加入savage试剂10ml,其中,savage试剂中氯仿与正丁醇体积比为5:1,室温充分搅拌1h,离心,取上清液,重复9次,直至离心后液面中间无白色固体,上清液冷冻干燥,即得脱蛋白后的莼菜粗多糖3.3g,提取率11.0%;
67.实施例7:莼菜粗多糖的提取
68.1、称取实施例1所制得的莼菜干粉35g,向莼菜干粉中加入蒸馏水,料液比为1:50,调节ph值为4.5,加入底物质量1%的果胶酶,控制提取温度55℃,提取2h,90℃灭活10min,过滤去除莼菜干粉,滤液减压浓缩至原体积1/5;
69.3、向浓缩液中边搅拌边加入三倍体积的无水乙醇,4℃静置过夜,离心,得褐色沉淀物;
70.4、沉淀物以12ml蒸馏水充分溶解,冷冻干燥,即得莼菜粗多糖;
71.5、将莼菜粗多糖充分溶解于50ml蒸馏水中,加入savage试剂10ml,其中,savage试剂中氯仿与正丁醇体积比为5:1,室温充分搅拌1h,离心,取上清液,重复7次,直至离心后液面中间无白色固体,上清液冷冻干燥,即得脱蛋白后的莼菜粗多糖2.7g,提取率7.7%;
72.实施例8:莼菜粗多糖的提取
73.1、称取实施例1所制得的莼菜干粉35g,向莼菜干粉中加入蒸馏水,料液比为1:50,调节ph值为3.5,加入底物质量2%的果胶酶,控制提取温度55℃,提取3h,90℃灭活10min,过滤去除莼菜干粉,滤液减压浓缩至原体积1/5;
74.3、向浓缩液中边搅拌边加入三倍体积的无水乙醇,4℃静置过夜,离心,得褐色沉淀物;
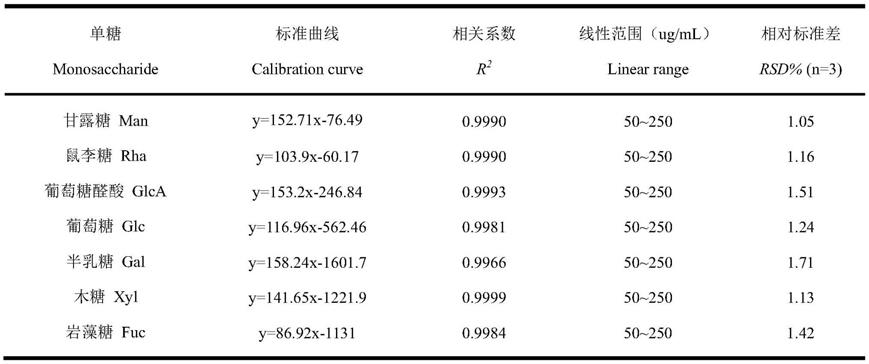
75.4、沉淀物以10ml蒸馏水充分溶解,冷冻干燥,即得莼菜粗多糖;
76.5、将莼菜粗多糖充分溶解于45ml蒸馏水中,加入savage试剂9ml,其中,savage试剂中氯仿与正丁醇体积比为5:1,室温充分搅拌1h,离心,取上清液,重复8次,直至离心后液面中间无白色固体,上清液冷冻干燥,即得脱蛋白后的莼菜粗多糖3.6g,提取率10.3%;
77.实施例9:莼菜粗多糖的提取
78.1、称取实施例1所制得的莼菜干粉20g,向莼菜干粉中加入蒸馏水,料液比为1:50,调节ph值为4.5,加入底物质量2%的果胶酶,控制提取温度65℃,提取1h,90℃灭活10min,过滤去除莼菜干粉,滤液减压浓缩至原体积1/5;
79.3、向浓缩液中边搅拌边加入三倍体积的无水乙醇,4℃静置过夜,离心,得褐色沉淀物;
80.4、沉淀物以10ml蒸馏水充分溶解,冷冻干燥,即得莼菜粗多糖;
81.5、将莼菜粗多糖充分溶解于40ml蒸馏水中,加入savage试剂8ml,其中,savage试剂中氯仿与正丁醇体积比为5:1,室温充分搅拌1h,离心,取上清液,重复6次,直至离心后液面中间无白色固体,上清液冷冻干燥,即得脱蛋白后的莼菜粗多糖1.9g,提取率9.5%;
82.实施例10:莼菜粗多糖的分离纯化
83.称取实施例1制备的脱蛋白后莼菜粗多糖200mg,充分溶解于4ml蒸馏水中,离心,取上清液,一次性上样于deae-52纤维素柱中进行分离纯化,分别使用蒸馏水及浓度为0.1、0.2、0.3、0.4、0.5mol/l的nacl溶液梯度洗脱,控制流速1ml/min,每10min收集一管,采用苯酚-硫酸法隔管检测洗脱液中多糖含量,绘制洗脱曲线,合并曲线中呈单一峰的洗脱组分,50℃减压旋转浓缩,经3500da透析袋蒸馏水透析72h,冷冻干燥,即得20.4mg目标酸性莼菜多糖bsp-5。
84.实施例11:酸性莼菜多糖bsp-5结构鉴定
85.(1)bsp-5成分测定
86.以实施例10所制备的目标酸性莼菜多糖bsp-5为样品,苯酚-硫酸法测定bsp-5总糖含量,硫酸-间羟联苯法测定bsp-5糖醛酸含量,考马斯蓝亮染色法测定bsp-5蛋白质含量,紫外全波长扫描测定bsp-5是否含有核酸。结果表明,bsp-5的总糖含量为93.25%,并含有30.91%的糖醛酸,不含有蛋白质。
87.由图2所示,紫外全波长扫描结果显示,bsp-5在280nm和260nm波长处没有特征吸收,这表明bsp-5不含有核酸,也再次证明bsp-5不含有蛋白质。
88.(2)bsp-5均一性及分子量测定
89.以实施例10所制备的目标酸性莼菜多糖bsp-5为样品,采用高效凝胶过滤色谱联合多角度激光散射法(hpsec-malls)测定bsp-5均一性及分子量。色谱柱为shodex ohpak sb-803hq(300mm
×
8.0mm)串联shodex ohpak sb-806hq(300mm
×
8.0mm),洗脱液为0.05m na2so4(含有0.02%nan3),柱温40℃,流速1ml/min,进样量500ul,样品过0.22μm滤膜过滤。
90.如图3所示,bsp-5在gpc色谱图中呈单一且对称的峰,表明bsp-5为一种均质多糖。其重均分子量mw为119.5kda,数均分子量mn为90.36kda,分散系数mw/mn为1.323。
91.(3)bsp-5单糖组成测定
92.精密称取实施例10所制备的目标酸性莼菜多糖bsp-5样品2mg于密封试管中,加入6ml浓度为2mol/l的三氟乙酸溶液,检漏合格后,110℃油浴锅中水解2h。反应结束后冷却至室温,水解液50℃减压旋转蒸干,加入3ml甲醇,再次蒸干,重复5次,以除去多余的三氟乙酸。残渣以1ml蒸馏水溶解得样品溶液,备用。
93.向上述样品溶液中加入1ml pmp甲醇溶液(0.5m)和1ml naoh(0.3m)溶液,混合均匀,密封,检漏合格后,70℃下反应1h。反应结束后冷却至室温,加入1ml hcl(0.3m)中和,加入5ml氯仿萃取,取水相,重复5次,水相过0.45μm滤膜过滤,备用。
94.紧密称取2mg各单糖标准品:d-半乳糖、d-甘露糖、l-鼠李糖、d-葡萄糖、d-木糖、l-岩藻糖和d-葡萄糖醛酸,按上述相同的方法进行pmp衍生化。
95.采用hplc进行分析,色谱条件:安捷伦1260hplc配备zorbax sb c18(4.6mm
×
250mm)柱;流动相:水和乙腈(80:20,v/v);流速:1ml/min;柱温:30℃;进样量:20μl;检测波长:245nm。根据各单糖标准品的标准曲线(峰面积-浓度)计算bsp-5单糖组成的摩尔比。
96.如图4所示,通过对比标准单糖及bsp-5的hplc色谱图可知,bsp-5主要由d-甘露糖、d-半乳糖、d-木糖和l-岩藻糖组成,如表1所示,将峰面积代入对应单糖的标准曲线,计算可知,其单糖组成摩尔比为0.08:1.39:1:0.23。由此可见,bsp-5是一种以半乳糖和木糖为主要糖成分的酸性杂质多糖。
97.表1 各单糖标准曲线
[0098][0099]
(4)bsp-5红外分析
[0100]
以实施例10所制备的目标酸性莼菜多糖bsp-5为样品,采用kbr压片法测定bsp-5的红外吸收光谱:称取充分干燥后的bsp-5样品1~2mg和光谱纯kbr粉末200mg,共同放置于干燥的玛瑙研钵中并充分研磨均匀,用压片机压片,以kbr为对照,用傅立叶红外光谱仪扫
描450~4000cm-1
范围bsp-5的红外光谱。
[0101]
如图5所示,在3419.56cm-1
和2925.81cm-1
处的强吸收峰为o-h键伸缩振动和c-h键伸缩振动,这两个峰是糖的典型吸收峰。在1730cm-1
左右有一个弱吸收峰,为c=o键的伸缩振动,表明bsp-5含有糖醛酸。在1608.52cm-1
处的吸收峰为o-h键的变形振动。在1417.58cm-1
处的吸收峰为c-h键的变形振动。在1445.64、1072.35和1029.92cm-1
处的吸收峰对应于吡喃糖型糖引起的非对称c-o-c键伸缩振动,表明bsp-5为吡喃糖。在838.98cm-1
处的吸收峰表明bsp-5为α构型。综上可知,bsp-5为α构型的吡喃糖。
[0102]
实施例12:体外抗氧化活性测定
[0103]
以实施例10所制备的目标酸性莼菜多糖bsp-5为样品,测定其体外抗氧化活性。
[0104]
1、dpph自由基清除率测定
[0105]
精密称取dpph 10mg,无水乙醇溶解并定容至50ml容量瓶中,得浓度为0.5mmol/l的dpph溶液,避光保存,备用。
[0106]
移取不同浓度bsp-5溶液(1、2、3、4、5mg/ml)3ml于15ml具塞试管中,加入1ml dpph溶液,充分混合,室温避光反应30min,517nm波长处测定吸光度值,以vc作为阳性对照,按下列公式计算dpph自由基清除率。dpph自由基清除率(%)=[1-(as-ab)/ac]
×
100%
[0107]
公式中,as—3ml样品溶液+1ml dpph溶液
[0108]
ab—3ml样品溶液+1ml无水乙醇
[0109]
ac—3ml无水乙醇+1ml dpph溶液
[0110]
实验重复三次,结果取平均值。如图6所示,莼菜酸性多糖bsp-5对dpph自由基的清除率随着多糖浓度的增加而上升,表现出一定的浓度依赖关系,其半数抑制浓度为1.68mg/ml,当浓度达到5mg/ml时,莼菜多糖对dpph自由基的清除率达到最高的73.89%,但低于同浓度下的vc,由此可以看出bsp-5对dpph自由基具有很高的清除能力。
[0111]
2、羟基自由基清除率测定
[0112]
采用fenton法测定羟基自由基清除率,于15ml具塞试管中加入不同浓度bsp-5溶液(1、2、3、4、5mg/ml)1ml,1ml feso4溶液(9mmol/l),1ml h2o2溶液(8.8mmol/l),1ml水杨酸-乙醇溶液(9mmol/l),摇匀,室温避光反应30min,510nm波长处测定吸光度值,以vc作为阳性对照,按下列公式计算羟基自由基清除率。
[0113]
羟基自由基清除率(%)=[1-(as-ab)/ac]
×
100%
[0114]
公式中,as—1ml样品溶液+1ml 1ml feso4溶液+1ml h2o2溶液+1ml水杨酸-乙醇溶液
[0115]
ab—1ml样品溶液+1ml feso4溶液+1ml蒸馏水+1ml水杨酸-乙醇溶液
[0116]
ac—1ml蒸馏水+1ml 1ml feso4溶液+1ml h2o2溶液+1ml水杨酸-乙醇溶液
[0117]
实验平行三次,结果取平均值。如图7所示,莼菜酸性多糖bsp-5对羟基自由基的清除率与多糖浓度表现出明显的浓度依赖关系,其半数抑制浓度为2.28mg/ml,当浓度达到5mg/ml时,莼菜多糖对羟基自由基的清除率达到最高的64.92%,但低于同浓度下的vc,由此可以看出bsp-5对羟基自由基具有良好的清除能力。
[0118]
3、abts
+
自由基清除率测定
[0119]
取7mmol/l abts
+
溶液0.2ml和2.6mmol/l过硫酸钾溶液0.2ml于10ml具塞试管中,充分混合,室温避光反应12-16h。使用前用ph=7.4的磷酸盐缓冲溶液稀释至734nm波长处
的吸光度为0.700
±
0.020。
[0120]
于15ml具塞试管中将1ml不同浓度bsp-5溶液(1、2、3、4、5mg/ml)与1ml稀释后abts
+
溶液充分混合,室温避光反应20min,734nm波长处测定吸光度值,以vc作为阳性对照,按下列公式计算abts
+
自由基清除率。abts
+
自由基清除率(%)=[1-(as-ab)/ac]
×
100%
[0121]
公式中,as—1ml样品溶液+1ml abts
+
溶液
[0122]
ab—1ml样品溶液+1ml蒸馏水
[0123]
ac—1ml蒸馏水+1ml abts
+
溶液
[0124]
实验重复三次,清除率取平均值。结果如图8所示,随着多糖浓度从1mg/ml增加至5mg/ml,abts
+
自由基的清除率不断上升,其半数抑制浓度为2.08mg/ml,这表明其清除率与多糖浓度为浓度依赖关系,当浓度增加至5mg/ml时,abts
+
自由基的清除率达到最高的86.38%,该浓度下vc的清除率为96.76%,由此可以看出bsp-5对abts
+
自由基具有显著的清除能力。
[0125]
4、fe
2+
离子螯合能力测定
[0126]
于15ml具塞试管中加入1ml不同浓度bsp-5溶液(1、2、3、4、5mg/ml),然后加入0.1ml fecl2(2mmol/l)与0.2ml菲咯嗪(5mmol/l)溶液,最后加入3.7ml蒸馏水,混合均匀,室温下反应10min,562nm波长处测定吸光度值,以edta-2na作为阳性对照,按下列公式计算fe
2+
离子螯合能力。
[0127]
fe
2+
离子螯合率(%)=[1-(as-ab)/ac]
×
100%
[0128]
公式中,as—1ml样品溶液+0.1ml fecl2溶液+0.2ml菲咯嗪溶液
[0129]
ab—1ml样品溶液+0.1ml fecl2溶液+0.2ml蒸馏水
[0130]
ac—1ml蒸馏水+0.1ml fecl2溶液+0.2ml菲咯嗪溶液
[0131]
实验重复三次,结果取平均值。如图9所示,随着多糖浓度的增加使得样品对fe
2+
离子的螯合率不断上升,其半数螯合浓度为1.80mg/ml,这说明其螯合率与多糖浓度为浓度依赖关系,当浓度增加至5mg/ml时,fe
2+
离子的螯合率达到66.87%,浓度下的edta-2na的螯合率为92.86%,由此可见bsp-5是一种优良的fe
2+
离子螯合剂。
[0132]
实施例13:肿瘤细胞增殖抑制活性测定
[0133]
以实施例10所制备的目标酸性莼菜多糖bsp-5为样品测定其对肿瘤细胞增殖抑制作用,以人肝癌细胞hepg2、人宫颈癌细胞hela和人前列腺癌细胞22rv1为体外研究对象,采用mtt法检测不同浓度bsp-5多糖对上述肿瘤细胞的增殖抑制作用,进而判断bsp-5多糖体外抗肿瘤活性。
[0134]
具体操作如下:
[0135]
用dmem无血清培养基分别配制浓度为25、50、100、200、300、400、500ug/ml的bsp-5多糖溶液,备用。
[0136]
将处于对数生长期的hepg2、hela、22rv1肿瘤细胞接种于96孔板上,每孔的细胞接种密度为5
×
103(100μm),于37℃、5%co2细胞培养箱中培养12h,使细胞充分贴壁,然后加入100μm不同浓度的bsp-5多糖溶液,每个浓度设置6个复孔,于37℃、5%co2细胞培养箱中作用24、48和72h。结束后,每孔中加入mtt溶液20ul,(5mg/ml),再在细胞培养箱中培养4h,最后,去除培养基,加入150ul dmso溶液,充分溶解甲赞晶体,570nm处测定吸光度,以细胞增殖抑制率来判断抗肿瘤活性,按下列公式计算细胞存活率。
[0137]
细胞增殖抑制率(%)=1-a
sample
/a
control
×
100%
[0138]
公式中,a
sample
—样品吸光度值
[0139]acontrol
—无血清培养基代替多糖样品溶液
[0140]
bsp-5对hepg2肿瘤细胞增殖抑制作用实验结果(n=3)
[0141]
由图10可知,给药24h后,当bsp-5的浓度增加到200ug/ml时,开始出现对hepg2肿瘤细胞增殖的抑制作用,随着给药浓度的增加,存活率也随之减小(p<0.05),这说明hepg2肿瘤细胞增殖抑制率具有浓度依赖性,当药物浓度达到500ug/ml时,细胞存活率达到最低的52.21%,与对照组相比,具有显著性差异(p<0.01)。增加给药时间,细胞存活率也呈现出降低趋势。给药48h后,bsp-5浓度为100ug/ml时就开始出现抑制作用,随着浓度的增加,各浓度下的细胞存活率与24h相比,均有显著降低(p<0.05),当药物浓度达到500ug/ml时,细胞存活率达到最低的46.03%,这说明hepg2肿瘤细胞增殖抑制率同时具有时间依赖性,当给药72h后,细胞存活率降低趋势开始减缓,各浓度下的细胞存活率与48h相比,无显著性差异(p>0.05)。
[0142]
bsp-5对hela肿瘤细胞增殖抑制作用实验结果(n=3)
[0143]
由图11可知,给药24h后,当bsp-5的浓度增加到50ug/ml时,开始出现对hela肿瘤细胞增殖的抑制作用,当药物浓度达到100ug/ml时,细胞存活率达到最低的72.31%,与对照组相比,具有显著性差异(p<0.01),然而增加药物浓度细胞存活率却没有明显变化(p>0.05),这说明hela肿瘤细胞增殖抑制率没有明显的浓度依赖性。给药48h后,100ug/ml药物浓度下的细胞存活率显著降低(p<0.05),且达到最低的63.13%,给药72h后,该药物浓度下的细胞存活率继续降低(p<0.05),达到58.07%,这说明hela肿瘤细胞增殖抑制率具有一定的时间依赖性,最佳作用浓度为100ug/ml。
[0144]
bsp-5对22rv1肿瘤细胞增殖抑制作用实验结果(n=3)
[0145]
由图12可知,给药24h后,当bsp-5的浓度从50ug/ml开始增加时,细胞存活率变化不明显(p>0.05),当药物浓度达到500ug/ml时,细胞存活率达到最低的80.35%,这说明22rv1肿瘤细胞增殖抑制率没有明显的浓度依赖性。给药48h后,细胞存活率与24h相比,总体呈现下降趋势,当药物浓度为500ug/ml时,细胞存活率达到最低的69.36%,与给药24h相比,显著降低(p<0.05)。给药72h后,细胞存活率下降趋势大幅度减缓,当药物浓度为500ug/ml时,细胞存活率达到最低的63.86%,这说明22rv1肿瘤细胞增殖抑制率具有一定的时间依赖性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1