陈皮陈化度的量化检测方法与流程
1.本发明涉及中药质量检测技术领域,特别涉及一种陈皮陈化度的量化检测方法。
背景技术:
2.传统中医理论认为陈皮“陈久者良”,其中“陈久者”指的是“有效陈化”时间久的陈皮(即陈化年份高的陈皮),而非“存皮时长”的陈皮(即储藏年份高的陈皮);经过有效的陈化后,陈皮的内在成分能够进行变化,去除温燥之性,且增强理气健脾,燥湿化痰之功,呈现出越陈越香,价值越高的现象,这才有“一两陈皮一两金,百年陈皮赛黄金”的说法。
3.行业标准《db44t 604-2009地理标志产品新会陈皮》阐述了陈皮三种的不同年份概念,即生产年份、历史年份和陈化年份;其中:
①
生产年份,指采收果实并加工成果皮的年份,说明样品什么年份生产;
②
历史年份(又称“储藏年份”),指以检验年份减去生产年份加一年计算出的年份,说明样品有多久;
③
陈化年份,指以年份表示的陈皮陈化度,说明样品达到相应等级的陈化水平。通过该标准对陈化年份与储藏年份两个时间概念的定义,可知两者的关系——储藏年份低的陈皮,其陈化年份也一定低;储藏年份高的陈皮,其陈化年份却不一定高,只有陈皮在储藏过程中得到有效的陈化,其储藏年份才接近陈化年份。同时,该标准还定义了其他专业术语,如:“陈化”:在自然干爽通风的条件下(注:在此条件下储藏的陈皮,称为“自然陈化陈皮”),产品贮存在透气性良好的包装容器内,随着时间变化,干柑皮其有效内含物的消长变化而导致其色、香、味和成分变化的过程。“陈化度”:用年份表示的新会陈皮不同时期的感官性状。该标准虽然提出了陈化度的概念,但没有提及如何客观量化表达的方法,且只涉及陈皮的感官性状,具有一定的局限性。同时,对于陈皮具体陈化时间,历代本草书籍说法不一,《药鉴》载有“陈皮须用隔年陈”;《本草要略》云“陈皮隔年者方可用”;《日用本草》载:“陈皮多年者更妙”;即使是同一古籍,对于陈皮陈化时间尚未明确。此外,陈皮陈化速度的快慢,受储藏温度、储藏湿度以及透气性不同的存储容器等因素的影响。依靠经验、通过感官性状等传统评价手段难以从量上判别陈皮的陈化度。
4.如何实现陈化度的量化检测,对陈皮的质量判断至关重要。
技术实现要素:
5.基于此,本发明的目的包括提供一种陈皮陈化度的量化检测方法,实现对陈皮陈化度的量化检测,为判断陈皮的陈化年份提供依据。
6.一种陈皮陈化度的量化检测方法,所述量化检测方法包括如下步骤:
7.构建陈化度值与陈皮成分含量的函数式;其中,所述陈皮成分包含根皮酚-β-d-葡糖苷、香草酸、柠檬烯和γ-萜品烯,所述函数式如下:
[0008][0009]
式中,chd为陈化度值,d1为香草酸含量,d2为根皮酚-β-d-葡糖苷含量,h1为柠檬烯在陈皮挥发油中的相对含量,h2为γ-萜品烯在陈皮挥发油中的相对含量;
[0010]
分别检测陈皮样品中根皮酚-β-d-葡糖苷和香草酸的含量以及挥发油中柠檬烯和γ-萜品烯的相对含量,并将所得检测结果带入所述函数式,根据所得陈化度值确定所述陈皮样品对应的陈化度。
[0011]
在本发明的一些实施方式中,检测所述的香草酸和/或根皮酚-β-d-葡糖苷的含量采用超高效液相色谱法,所述超高效液相色谱法的条件包括:
[0012]
固定相:c18色谱柱;
[0013]
流动相:流动相a为甲醇,流动相b为磷酸体积百分比为0.08%-0.12%的磷酸水溶液;
[0014]
采用梯度洗脱的方式,所述梯度洗脱的程序包括:0min-3min,所述流动相a的体积百分比为3%;3min-5min,所述流动相a的体积百分比为由3%上升至5%;5min-7min,所述流动相a的体积百分比由5%上升至20%;7min-8min,所述流动相a的体积百分比为20%;8min-17min,所述流动相a的体积百分比由20%上升至30%;17min-19min,所述流动相a的体积百分比由30%上升至50%;19min-20min,所述流动相a的体积百分比为50%。
[0015]
在本发明的一些实施方式中,所述超高效液相色谱法的条件还包括:流速:0.45ml/min-0.55ml/min;或/和,柱温:25℃-35℃;或/和,进样量:1.5μl-2.5μl;或/和,检测波长:210nm-220nm,255nm-265nm。
[0016]
在本发明的一些实施方式中,所述检测波长为220nm,260nm.
[0017]
在本发明的一些实施方式中,检测所述陈皮样品中所述的根皮酚-β-d-葡糖苷和香草酸的含量的过程中,对应的供试品溶液的制备步骤包括:取所述陈皮样品,提取溶剂提取,收集提取液,制备样品溶液。
[0018]
在本发明的一些实施方式中,所述提取溶剂包含甲醇体积百分比为25%-35%的甲醇水溶液。
[0019]
在本发明的一些实施方式中,提取的方式为超声提取,超声提取的时间为25min-35min。
[0020]
在本发明的一些实施方式中,每1g所述陈皮样品对应的所述提取溶剂的用量为40ml-50ml。
[0021]
在本发明的一些实施方式中,收集所述提取液的方式为离心,离心的转速为8000r
·
min-1-12000r
·
min-1
,离心的时间为8min-12min。
[0022]
在本发明的一些实施方式中,检测所述挥发油中柠檬烯和γ-萜品烯的相对含量采用气质联用法,所述气质联用法的条件包括:
[0023]
固定相:hp-5ms色谱柱;
[0024]
载气:氦气;
[0025]
采用程序升温模式,所述程序升温模式的程序包括:柱初始温度为50℃,保持2min;以3℃/min的速率升至70℃,保持10min;以8℃/min的速率升至110℃,保持5min;以4℃/min的速率升至210℃,保持2min。
[0026]
在本发明的一些实施方式中,所述气质联用法的条件还包括:流速:0.8ml/min-1.2ml/min。
[0027]
在本发明的一些实施方式中,所述气质联用法的条件还包括:进样口温度:225℃-235℃。
[0028]
在本发明的一些实施方式中,所述气质联用法的条件还包括:分流比:(45-55):1。
[0029]
在本发明的一些实施方式中,所述气质联用法的条件还包括:离子源温度:225℃-235℃。
[0030]
在本发明的一些实施方式中,所述气质联用法的条件还包括:电子能量:35ev-75ev。
[0031]
在本发明的一些实施方式中,所述气质联用法的条件还包括:离子采集范围35m/z-450m/z。
[0032]
在本发明的一些实施方式中,检测所述陈皮样品中所述柠檬烯和γ-萜品烯的相对含量的过程中,对应的所述陈皮样品的前处理方法包括如下步骤:将所述陈皮样品置于密封条件下平衡处理,固相顶空萃取,解吸。
[0033]
在本发明的一些实施方式中,平衡处理的温度为85℃-95℃,平衡处理的时间为8min-12min。
[0034]
在本发明的一些实施方式中,固相顶空萃取采用car/dvb/pdms萃取头。
[0035]
在本发明的一些实施方式中,固相顶空萃取的时间为35min-45min。
[0036]
在本发明的一些实施方式中,解吸的温度为240℃-260℃,解吸的时间为2.5min-3.5min。
[0037]
在本发明的一些实施方式中,根据所得陈化度值确定所述陈皮样品对应的陈化度包括:所述陈皮样品的陈化度与所述陈化度值呈正相关关系,所述陈化度值越大,所述陈皮样品对应的所述陈化度越高。
[0038]
与传统技术相比,本发明具备如下有益效果:
[0039]
本发明的发明人在对不同陈化时间、不同存储容器的广陈皮样品的研究中发现了含量与陈皮的陈化度呈“消”“长”关系的成分(包括根皮酚-β-d-葡糖苷、香草酸、柠檬烯和γ-萜品烯),并基于这种“消”“长”关系建立了其与陈化度值的函数模型,能用于测定陈化度的大小,可直接用于判断陈皮样品是否得到有效的陈化以及陈化年份的高低,为陈皮质量检测提供量化手段。同时,本发明首次用客观数据证实了“陈皮在陈化过程中,存储容器对其陈化的影响大于储藏时间的”,为地方标准《db44/t 604-2009地理标志产品新会陈皮》中“陈皮需陈化三年及以上”的规定提供了科学解释。
[0040]
本发明创新性的提出基于消长变化成分含量比值与挥发油成分相对含量比值的“陈化度”评价理论,构建首个量化陈皮陈化度的公式,突破了以储藏年份论陈皮品质高低的传统模式,实现对陈皮陈化度的量化检测,丰富和完善了现有陈皮品质评价模式,并可用于陈皮品质等级的划分。同时,为半夏、麻黄、吴茱萸、枳壳、狼毒等其他“陈药”的质量研究提供了新的研究思路与方法。
附图说明
[0041]
为了更清楚地说明本技术实施例中的技术方案、更完整地理解本技术及其有益效果,下面将对实施例描述中所需要使用的附图作简单的介绍。显而易见地,下面描述中的附图仅仅是本技术的一些实施例,对本领域技术人员来说,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0042]
图1为不同存储容器的陈皮样品hplc色谱图(220nm及260nm);
[0043]
图2为成分1 1
h-nmr图;
[0044]
图3为成分1 13
c-nmr图;
[0045]
图4为成分1质谱图;
[0046]
图5为成分1紫外吸收光谱图;
[0047]
图6为成分2 1
h-nmr图;
[0048]
图7为成分2 13
c-nmr图;
[0049]
图8为成分2质谱图;
[0050]
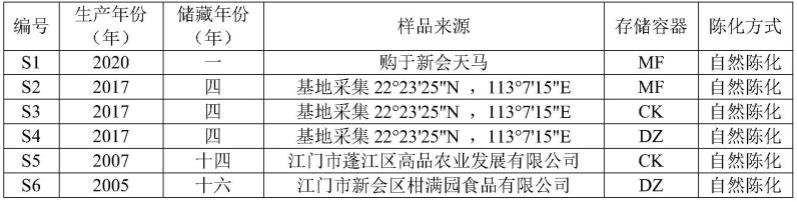
图9为成分2紫外吸收光谱图;
[0051]
图10为成分3 1
h-nmr图;
[0052]
图11为成分3 13
c-nmr图;
[0053]
图12为成分3质谱图;
[0054]
图13为成分3紫外吸收光谱图;
[0055]
图14为混合标准品紫外吸收光谱图;
[0056]
图15为陈皮样品目标色谱峰紫外吸收光谱图;
[0057]
图16为陈皮样品与标准品在220nm及260nm波长下色谱图;
[0058]
图17为三种标准品的标准曲线图;
[0059]
图18为陈皮样品gc/ms总离子流图(tic);
[0060]
图19为试验样品中成分含量比值与陈化度的变化趋势图;
[0061]
图20为验证样品中成分含量比值与陈化度的变化趋势图;
[0062]
图21本发明的技术路线图。
具体实施方式
[0063]
下面结合附图、实施方式和实施例,对本发明作进一步详细的说明。应理解,这些实施方式和实施例仅用于说明本发明而不用于限制本发明的范围,提供这些实施方式和实施例的目的是使对本发明公开内容理解更加透彻全面。还应理解,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式和实施例,本领域技术人员可以在不违背本发明内涵的情况下作各种改动或修改,得到的等价形式同样落于本技术的保护范围。此外,在下文的描述中,给出了大量具体的细节以便提供对本发明更为充分地理解,应理解,本发明可以无需一个或多个这些细节而得以实施。
[0064]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述实施方式和实施例的目的,不是旨在于限制本发明。
[0065]
本发明提供一种陈皮陈化度的量化检测方法,所述量化检测方法包括如下步骤:
[0066]
构建陈化度值与陈皮成分含量的函数式;其中,所述陈皮成分包含根皮酚-β-d-葡糖苷、香草酸、柠檬烯和γ-萜品烯,所述函数式如下:
[0067][0068]
式中,chd为陈化度值,d1为香草酸含量,d2为根皮酚-β-d-葡糖苷含量,h1为柠檬烯在陈皮挥发油中的相对含量,h2为γ-萜品烯在陈皮挥发油中的相对含量;
[0069]
分别检测陈皮样品中根皮酚-β-d-葡糖苷和香草酸的含量以及挥发油中柠檬烯和γ-萜品烯的相对含量,并将所得检测结果带入所述函数式,根据所得陈化度值确定所述陈皮样品对应的陈化度。
[0070]
在其中一个示例中,检测所述的香草酸和/或根皮酚-β-d-葡糖苷的含量采用超高效液相色谱法,所述超高效液相色谱法的条件包括:固定相:c18色谱柱;流动相:流动相a为甲醇,流动相b为磷酸体积百分比为0.08%-0.12%的磷酸水溶液;采用梯度洗脱的方式,所述梯度洗脱的程序包括:0min-3min,所述流动相a的体积百分比为3%;3min-5min,所述流动相a的体积百分比为由3%上升至5%;5min-7min,所述流动相a的体积百分比由5%上升至20%;7min-8min,所述流动相a的体积百分比为20%;8min-17min,所述流动相a的体积百分比由20%上升至30%;17min-19min,所述流动相a的体积百分比由30%上升至50%;19min-20min,所述流动相a的体积百分比为50%。
[0071]
在其中一个示例中,所述超高效液相色谱法的条件还包括:流速:0.45ml/min-0.55ml/min。例如0.45ml/min、0.46ml/min、0.47ml/min、0.48ml/min、0.49ml/min、0.50ml/min、0.51ml/min、0.52ml/min、0.53ml/min、0.54ml/min、0.55ml/min。
[0072]
在其中一个示例中,所述超高效液相色谱法的条件还包括:柱温:25℃-35℃。例如柱温为25℃、26℃、27℃、28℃、29℃、30℃、31℃、32℃、33℃、34℃、35℃。
[0073]
在其中一个示例中,所述超高效液相色谱法的条件还包括:进样量:1.5μl-2.5μl。例如进样量为1.5μl、1.6μl、1.7μl、1.8μl、1.9μl、2.0μl、2.1μl、2.2μl、2.3μl、2.4μl、2.5μl
[0074]
在其中一个示例中,所述超高效液相色谱法的条件还包括:检测波长:210nm-220nm(例如为210nm、211nm、212nm、213nm、214nm、215nm、216nm、217nm、218nm、219nm、220nm),255nm-265nm(例如为255nm、256nm、257nm、258nm、259nm、260nm)。优选地,检测波长为:220nm,260nm。
[0075]
优选地,所述超高效液相色谱法的条件包括:固定相:c18色谱柱;流动相:流动相a为甲醇,流动相b为磷酸体积百分比为0.08%-0.12%的磷酸水溶液;采用梯度洗脱的方式,所述梯度洗脱的程序包括:0min-3min,所述流动相a的体积百分比为3%;3min-5min,所述流动相a的体积百分比为由3%上升至5%;5min-7min,所述流动相a的体积百分比由5%上升至20%;7min-8min,所述流动相a的体积百分比为20%;8min-17min,所述流动相a的体积百分比由20%上升至30%;17min-19min,所述流动相a的体积百分比由30%上升至50%;19min-20min,所述流动相a的体积百分比为50%;流速:0.45ml/min-0.55ml/min;柱温:25℃-35℃;进样量:1.5μl-2.5μl;检测波长:210nm-220nm,255nm-265nm。
[0076]
在其中一个示例中,检测所述陈皮样品中所述的根皮酚-β-d-葡糖苷和香草酸的含量的过程中,对应的供试品溶液的制备步骤包括:取所述陈皮样品,提取溶剂提取,收集提取液,制备样品溶液。
[0077]
在其中一个示例中,所述提取溶剂包含甲醇体积百分比为25%-35%的甲醇水溶液,例如甲醇体积百分比为25%、26%、27%、28%、29%、30%、31%、32%、33%、34%、35%。
[0078]
在其中一个示例中,提取的方式为超声提取,超声提取的时间为25min-35min,例如25min、26min、27min、28min、29min、30min、31min、32min、33min、34min、35min。
[0079]
在其中一个示例中,每1g所述陈皮样品对应的所述提取溶剂的用量为40ml-50ml,
例如40ml、42ml、44ml、46ml、48ml、50ml。
[0080]
在其中一个示例中,收集所述提取液的方式为离心,离心的转速为8000r
·
min-1-12000r
·
min-1
,离心的时间为8min-12min。离心的转速例如为:8000r
·
min-1
、8500r
·
min-1
、9000r
·
min-1
、10000r
·
min-1
、11000r
·
min-1
、12000r
·
min-1
。
[0081]
在其中一个示例中,所述提取溶剂包含甲醇体积百分比为25%-35%的甲醇水溶液;提取的方式为超声提取,超声提取的时间为25min-35min;每1g所述陈皮样品对应的所述提取溶剂的用量为40ml-50ml;收集所述提取液的方式为离心,离心的转速为8000r
·
min-1-12000r
·
min-1
,离心的时间为8min-12min。
[0082]
在其中一个示例中,检测所述挥发油中柠檬烯和γ-萜品烯的相对含量采用气质联用法,所述气质联用法的条件包括:固定相:hp-5ms色谱柱;载气:氦气;采用程序升温模式,所述程序升温模式的程序包括:柱初始温度为50℃,保持2min;以3℃/min的速率升至70℃,保持10min;以8℃/min的速率升至110℃,保持5min;以4℃/min的速率升至210℃,保持2min。
[0083]
在其中一个示例中,所述气质联用法的条件还包括:流速:0.8ml/min-1.2ml/min,例如0.8ml/min、0.85ml/min、0.9ml/min、0.95ml/min、1ml/min、1.1ml/min、1.2ml/min。
[0084]
在其中一个示例中,所述气质联用法的条件还包括:进样口温度:225℃-235℃。例如为225℃、227℃、229℃、230℃、231℃、233℃、235℃。
[0085]
在其中一个示例中,所述气质联用法的条件还包括:分流比:(45-55):1。例如45:1、46:1、47:1、48:1、49:1、50:1、51:1、52:1、53:1、54:1、55:1。
[0086]
优选地,所述气质联用法的条件还包括:流速:0.8ml/min-1.2ml/min;进样口温度:225℃-235℃;分流比:(45-55):1。
[0087]
在其中一个示例中,所述气质联用法的条件还包括:离子源温度:225℃-235℃。
[0088]
在其中一个示例中,所述气质联用法的条件还包括:电子能量:35ev-75ev。
[0089]
在其中一个示例中,所述气质联用法的条件还包括:离子采集范围35m/z-450m/z。
[0090]
优选地,所述气质联用法的条件还包括:离子源温度:225℃-235℃;所述气质联用法的条件还包括:电子能量:35ev-75ev;离子采集范围35m/z-450m/z。
[0091]
在其中一个示例中,检测所述陈皮样品中所述柠檬烯和γ-萜品烯的相对含量的过程中,对应的所述陈皮样品的前处理方法包括如下步骤:将所述陈皮样品置于密封条件下平衡处理,固相顶空萃取,解吸。
[0092]
在其中一个示例中,平衡处理的温度为85℃-95℃,平衡处理的时间为8min-12min。平衡处理的温度例如为85℃、86℃、87℃、88℃、89℃、90℃、91℃、92℃、93℃。平衡处理的时间为8min、9min、10min、11min、12min。
[0093]
在其中一个示例中,固相顶空萃取采用car/dvb/pdms萃取头。
[0094]
在其中一个示例中,固相顶空萃取的时间为35min-45min。例如35min、36min、37min、38min、39min、40min、41min、42min、43min、44min、45min。
[0095]
在其中一个示例中,解吸的温度为240℃-260℃,解吸的时间为2.5min-3.5min。解吸的温度例如为240℃、245℃、250℃、255℃、260℃。解吸的时间为2.5min、2.6min、2.7min、min、2.8min、2.9min、3min、3.1min、3.2min、3.3min、3.4min、3.5min。
[0096]
优选地,平衡处理的温度为85℃-95℃,平衡处理的时间为8min-12min;固相顶空
萃取采用car/dvb/pdms萃取头;固相顶空萃取的时间为35min-45min;解吸的温度为240℃-260℃,解吸的时间为2.5min-3.5min。
[0097]
在其中一个示例中,根据所得陈化度值确定所述陈皮样品对应的陈化度包括:所述陈皮样品的陈化度与所述陈化度值呈正相关关系,所述陈化度值越大,所述陈皮样品对应的所述陈化度越高。
[0098]
在其中一个示例中,所述陈皮的陈化方式为自然陈化。也就说,本发明的检测方法,可以用于自然陈化陈皮的检测。用于自然陈化陈皮的检测的过程中,无法确认陈皮样品是非自然陈化陈皮还是自然陈化陈皮的情况下,可以根据需要,采用合适的方法先对陈皮样品进行鉴别,然后再用本发明的检测方法对鉴别出的自然陈化陈皮进行检测。因此,本发明的检测方法还可以进一步包括对陈皮样品进行鉴别的步骤,本发明对鉴别的方法不做特别限定。
[0099]
具体实施例
[0100]
下面将结合实施例对本发明的实施方案进行详细描述。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,优先参考本发明中给出的指引,还可以按照本领域的实验手册或常规条件,还可以按照制造厂商所建议的条件,或者参考本领域已知的实验方法。
[0101]
下述的具体实施例中,涉及原料组分的量度参数,如无特别说明,可能存在称量精度范围内的细微偏差。涉及温度和时间参数,允许仪器测试精度或操作精度导致的可接受的偏差。
[0102]
1仪器与试药
[0103]
1.1仪器
[0104]
waters 2695-2996液相色谱仪(美国沃特世公司)、kromasil 100-5-c18色谱柱(5μm,4.6mm
×
250mm,瑞典阿克苏诺贝尔公司)、制备型hplc系统汉邦dac-150(江苏汉邦科技有限公司)、分析型hplc系统汉邦np7001(江苏汉邦科技有限公司)、分析型hplc系统agela hp-q-p010z(天津博纳艾杰尔科技有限公司)、5l旋蒸机组bc-r501ca和20l旋蒸机组bc-r2012c(bc)(上海贝凯生物化工设备有限公司)、bruker超导脉冲傅立叶变换核磁共振波谱仪bruker av ii-600mhz,400mhz(瑞士bruker公司)、waters三重四极杆液质联用仪quattro premier xe(美国沃特世公司)、waters h-class uplc-pda detector超高效液相色谱(美国沃特世公司)、waters xselect csh c18色谱柱(2.5μm,3.0
×
100mm,美国沃特世公司)、xpr 26/a百万分之一电子天平(瑞士梅特勒-托利多公司)、sartorius bs420s万分之一电子天平、tgl-15b离心机(上海安亭科学仪器厂)、milli-q reference纯水机(法国millipore)、sb-5200dt超声波清洗仪(宁波新芝生物科技股份有限公司)、尼龙66 0.22μm微孔滤膜、wxj型粉碎机(上海凯旋中药机械制造有限公司)。
[0105]
安捷伦hp-5ms(30mm
×
0.25mm
×
0.25μm)色谱柱(美国agilent公司);7890a-5975c气相色谱-质谱联用仪(美国agilent公司):50/30μm pdms/dvb固相微萃取仪-纤维头(美国supelco公司,灰色),40ml spme专用采样瓶(美国supelco公司)。
[0106]
1.2试剂与试药
[0107]
5-羟甲基-2-呋喃甲酸标准品(纯度99.40%,cas:6338-41-6,上海施丹德标准技术服务有限公司);间苯三酚-o-葡萄糖苷(别名根皮酚-β-d-葡糖苷,纯度98.00%,cas:
28217-60-9,中山市成诺生物科技有限公司);香草酸(纯度99.80%,批号:110776-201503,中国食品药品检定研究院);维采宁-2标准品(纯度99.57%,批号:19121201,成都格利普生物科技有限公司);色谱级甲醇(德国merck)、超纯水、甲醇(广州化学试剂厂)。
[0108]
1.3样品信息
[0109]
本研究的试验样品主要来源为新会陈皮基地采集以及新会产区采购,编号为s1~s6,将获得的广陈皮样品采用三种不同的存储容器进行陈化(见表1),并放置于干燥通风的环境;样品来源信息,详见表2。
[0110]
以上样品经广州中医药大学中药鉴定教研室主任黄海波副教授鉴定为芸香科植物茶枝柑citrus reticulate
‘
chachi’的果皮。其中s2~s4为同一采集地、生产年份、成熟度、贮藏环境的同来源样品,分别储藏于不同的存储容器中。
[0111]
表1、陈皮样品三种存储容器
[0112]
序号存储容器透气性编码1麻袋装、网箱装等良好dz2塑料袋装敞口、纸箱装等一般ck3密封袋、玻璃罐密封等较差mf
[0113]
表2、陈皮样品信息表
[0114][0115]
2方法与结果
[0116]
2.1陈皮陈化过程中消长变化成分的含量及其比值研究
[0117]
陈皮在陈化过程中外观颜色会发生变化,尤其是内囊颜色的变化更明显;相关研究表明,中药颜色的细微差异往往也预示着其内在有效成分的差异。本研究选择陈化时间差异大的两个陈皮样品,比较两者之间的内在差异成分;然后对差异成分进行分离、鉴定,并购置对应的对照品,测定其含量;计算差异成分含量之间的比值,并分析比值与陈化年份之间的关联性;最后,将与陈化年份相关性强的比值纳入陈化度的公式中。
[0118]
2.1.1消长变化成分的鉴定
[0119]
为了探索如何量化陈化度并建立广陈皮陈化度模型,本研究选择陈化时间差异大的两个陈皮样品s1、s6进行研究,分离并鉴定两种陈皮样品之间的3个主要差异成分。运用多种色谱法进行分离纯化,并通过核磁共振和高分辨质谱技术,比对氢谱和碳谱数据、质谱数据、光谱数据,对3个化合物进行结构鉴定。
[0120]
2.1.1.1供试品及标准品溶液的制备
[0121]
供试品溶液的制备:将陈皮样品粉碎过三号筛,称约0.5g,置于具塞三角瓶加入30%(v/v)甲醇水溶液25ml,超声30min,放冷后以30%(v/v)甲醇水溶液补重,离心10分钟,取上清液,用0.22μm有机滤膜过滤,进样10μl。
[0122]
标准品溶液的制备:称取维采宁-2标准品适量,置于25ml容量瓶中,加入50%(v/
v)甲醇水溶液制成标准品溶液,储存于4℃冰箱中。
[0123]
2.1.1.2消长变化成分的确认
[0124]
根据项目组的前期研究成果,采用waters 2695-2996液相色谱仪,kromasil 100-5-c18色谱柱(5μm,4.6mm
×
250mm);以甲醇-0.1%(v/v)磷酸水为流动相进行梯度洗脱,洗脱程序:5%甲醇(0-5min),5~10%甲醇(5-10min),10%甲醇(10-15min),10~20%甲醇(15-25min),20~25%甲醇(25-40min),25~40%甲醇(35-50min),40%甲醇(50-55min),40~50%甲醇(55-60min),50%甲醇(60-65min),50~90%甲醇(65-66min),90%甲醇(66-72min);流速1ml
·
min-1
;柱温30℃;进样量:10μl;检测波长220nm、260nm。
[0125]
记录色谱图,见图1。
[0126]
基于研究组的前期研究,选取在陈化前后差异较为明显的三个差异峰(差异峰1、差异峰2、差异峰3)进行后续提纯分离并鉴定。
[0127]
2.1.1.3成分分离与鉴定
[0128]
(1)成分分离纯化
[0129]
取s1陈皮样品600g粉碎,用30%(v/v)甲醇水溶液超声提取4次,每次0.5h,过滤合并提取液过nm200柱层析,甲醇-水梯度洗脱,得到30%(v/v)甲醇水溶液洗液约16l,用50℃减压浓缩至约2.2l,aq-c18柱反复高压液相制备(甲醇:0.1%pa=4:96为流动相,波长:220nm),得到成分1(差异峰1成分)合格制备液。将合格制备液45℃减压浓缩至小体积,aq-c18富集,水洗脱酸后,90%(v/v)甲醇水溶液解析。90%(v/v)甲醇水溶液解析液45℃减压浓缩干,刮出40℃恒温鼓风干燥恒重后,真空干燥至恒重,得到类白色固体(成分1)约45mg。
[0130]
取s6陈皮样品510g粉碎,同法得30%(v/v)甲醇水溶液洗液约16l,用50℃减压浓缩至约10l,aq-c18柱反复高压液相制备(乙腈:0.1%pa=10:90为流动相,波长:260nm),得到成分2(差异峰2成分)合格制备液,45℃减压浓缩至小体积,aq-c18富集,水洗脱酸后,90%(v/v)甲醇水溶液解析,45℃减压浓缩干,刮出40℃恒温鼓风干燥恒重后,真空干燥至恒重,得到灰黄色固体(成分2)约50mg。
[0131]
取s6陈皮样品510g粉碎,同法得50%(v/v)甲醇水溶液洗液约10l,用50℃减压浓缩至约6l,c18柱反复高压液相制备(甲醇:0.1%pa=30:70为流动相,波长:260nm),得到成分3(差异峰3成分)合格制备液,45℃减压浓缩至小体积,aq-c18富集,水洗脱酸后,90%(v/v)甲醇水溶液解析,45℃减压浓缩干,刮出40℃恒温鼓风干燥恒重后,真空干燥至恒重,得到淡黄色固体(成分3)约24mg。
[0132]
(2)成分结构鉴定
[0133]
成分1为类白色固体(甲醇);1h-nmr(meod-d4,500mhz)δ:3.47-3.37(4h,m,h-2
′
,h-3
′
,h-4
′
,h-5
′
),3.71(1h,dd,j=12.0,4.5hz,h-6a
′
),3.89(1h,d,j=12.2hz,h-6b
′
),4.82(1h,d,j=7.5hz,h-1
′
),5.95(1h,t,j=2.0hz,h-4),6.08(2h,d,j=2.0hz,h-2,h-6),见图2;
13
c-nmr(meod-d4,150mhz)δ:161.0(c-1),96.8(c-2,c-6),160.3(c-3,c-5),98.1(c-4),102.2(c-1
′
),75.0(c-2
′
),78.1(c-3
′
),71.4(c-4
′
),78.2(c-5
′
),62.6(c-6
′
),见图3。
[0134]
质谱esi-ms m/z:289.0[m+h]+,311.0[m+na]+,287.1[m-h]-(见图4),紫外uv:λmax 220nm,268nm(见图5)。
[0135]
与间苯三酚-o-葡萄糖苷的紫外吸收图、分子量及分子结构均相符,确定成分1为间苯三酚-o-葡萄糖苷(别名:根皮酚-β-d-葡糖苷)。
[0136]
成分2为灰黄色固体(甲醇);1h-nmr(dmso-d6,400mhz)δ:12.94(bs,1h),7.13(1h,d,j=3.4hz),6.45(1h,d,j=3.4hz),5.45(1h,bs),4.44(2h,s),见图6;
13
c-nmr(dmso-d6,100mhz)δ:55.8(1c)109.0(1c),118.5(1c),144.0(1c),159.4(1c),159.7(1c),见图7。
[0137]
质谱esi-ms m/z:143.5[m+h]+,165.0[m+na]+,141.0[m-h]-(见图8);紫外uv:λmax 256nm(见图9)。
[0138]
与5-羟甲基-2-呋喃甲酸的紫外吸收图、分子量及分子结构均相符,确定成分2为5-羟甲基-2-呋喃甲酸。
[0139]
成分3为淡黄色固体(甲醇);1h-nmr(dmso-d6,600mhz)δ:7.43(1h,m,h-2),6.84(1h,d,j=7.9hz,h-5),7.44(1h,d,j=1.7hz,h-6),3.80(3h,s,3-ome),见图10;
13
c-nmr(dmso-d6,150mhz)δ:121.6(c-1),115.1(c-2),147.3(c-3),151.1(c-4),112.7(c-5),123.5(c-6),167.2(cooh),55.6(ome)见图11。
[0140]
质谱esi-ms m/z:169.1[m+h]+,206.9[m+k]+,166.9[m-h]-(见图12);紫外uv:λ
max 205nm,217nm,260nm,292nm(见图13)。
[0141]
与香草酸的紫外吸收图、分子量及分子结构均相符,确定成分3为香草酸。
[0142]
2.1.2消长变化成分的uplc含量测定
[0143]
由于上述被分离鉴定的3个成分在不同陈化时间的陈皮中含量悬殊,采用样品s2建立根皮酚-β-d-葡糖苷的含量测定方法,采用s4建立5-羟甲基-2-呋喃甲酸和香草酸的含量测定方法。
[0144]
2.1.2.1含量测定方法
[0145]
色谱条件:waters h-class uplc-pda detector超高效液相色谱,色谱柱为waters xcsh c18色谱柱(2.5μm,3.0mm
×
100mm),流速0.5ml/min;柱温30℃;进样量2μl;检测波长为220nm、260nm;以甲醇-0.1%(v/v)磷酸水溶液为流动相进行梯度洗脱,洗脱程序:3%甲醇(0-3min),3~5%甲醇(3-5min),5~20%甲醇(5-7min),20%甲醇(7-8min),20~30%甲醇(8-17min),30~50%甲醇(17-19min),50%甲醇(19-20min)。
[0146]
混合标准品溶液的制备:取根皮酚-β-d-葡糖苷标准品精密称定,30%(v/v)甲醇定容至0.2024mg/ml;取5-羟甲基-2-呋喃甲酸标准品精密称定,甲醇定容至0.2012mg/ml;取香草酸标准品精密称定,甲醇定容至0.2104mg/ml,标准品储备液均置于4℃冰箱中保存。分别精密量取上述标准品储备液置于容量瓶中,30%(v/v)甲醇定容,制成混合标准品。
[0147]
供试品溶液的制备:取陈皮样品粉末0.5g(过45目筛),精密称定,置50ml具塞锥形瓶中,加入25ml30%(v/v)甲醇,称重,超声处理30min,放冷后称重,并用30%(v/v)甲醇补足重量,10000r
·
min-1
离心10min,取上清液,用0.22μm微孔滤膜过滤,即得。
[0148]
计算方法:其中,
[0149]
c:三种成分的测得浓度(μg/ml);w:称样量(g)。
[0150]
2.1.2.2方法学考察
[0151]
由于上述3个成分在不同陈化程度的陈皮样品中的含量悬殊较大,为保证所建立定量方法的适用性,采用样品s2用于建立根皮酚-β-d-葡糖苷的含量测定方法,采用样品s4建立5-羟甲基-2-呋喃甲酸和香草酸的含量测定方法。专属性考察采用样品s4,其它方法学验证中采用样品s2检测根皮酚-β-d-葡糖苷,采用样品s4检测5-羟甲基-2-呋喃甲酸和香草酸。
[0152]
(1)专属性考察
[0153]
分别精密吸取标准品品溶液、s4供试品溶液各2μl,注入超高效液相色谱仪进行测定,在全波长扫描模式下记录目标峰的紫外吸收图,并记录在220nm、260nm波长下的色谱图。目标峰全波长扫描紫外吸收光谱图见图14和图15,220nm、260nm波长下色谱图见图16。样品色谱图中出现与对照品色谱图中保留时间一致(rt=2.23、7.13、11.76min)的色谱峰,目标色谱峰紫外吸收光谱图与标准品一致无干扰。
[0154]
(2)线性关系考察
[0155]
取根皮酚-β-d-葡糖苷标准品储备液加入30%(v/v)甲醇分别稀释成0.47、0.94、1.89、3.77、7.55、15.09、30.18、60.36μg
·
ml-1
的标准品溶液,取5-羟甲基-2-呋喃甲酸标准品储备液加入30%(v/v)甲醇分别稀释成0.07、0.16、0.32、0.63、1.27、2.53、5.06、10.12、20.24μg
·
ml-1
的标准品溶液,取香草酸标准品储备液加入30%(v/v)甲醇分别稀释成0.16、0.33、0.66、1.32、2.63、5.26、10.52、21.04μg
·
ml-1
的标准品溶液,经0.22μm微孔滤膜过滤后,取2μl注入超高效液相色谱仪中进行测定,对相应的色谱峰进行积分并计算标准曲线,标准曲线如图17。
[0156]
以对照品浓度x(μg
·
ml-1
)为横坐标,以峰面积y为纵坐标,绘制标准曲线,得根皮酚-β-d-葡糖苷回归方程为y=5.66
×
103x-3.81
×
102(r2=0.9999),浓度在0.47~60.36μg
·
ml-1
之间与峰面积值呈现良好的线性关系;5-羟甲基-2-呋喃甲酸回归方程为y=2.29
×
104x-1.61
×
103(r2=0.9999),浓度在0.07~20.24μg
·
ml-1
之间与峰面积值呈现良好的线性关系;香草酸回归方程为y=1.61
×
104x-1.49
×
103(r2=0.9999),浓度在0.16~21.04μg
·
ml-1
之间与峰面积值呈现良好的线性关系。
[0157]
(3)精密度考察
[0158]
取根皮酚-β-d-葡糖苷标准品(7.545μg
·
ml-1
)、5-羟甲基-2-呋喃甲酸标准品(1.265μg
·
ml-1
)、香草酸标准品(1.315μg
·
ml-1
)混合标准品,按“2.1.2.1”项下色谱条件进样分析,连续测定6次,记录峰面积,计算rsd值。
[0159]
结果如表3。三种成分的rsd均《1%,结果表明仪器精密度良好。
[0160]
表3、精密度考察
[0161][0162]
(4)重复性考察
[0163]
分别取陈皮样品s2(用于检测根皮酚-β-d-葡糖苷)及s4(用于检测5-羟甲基-2-呋喃甲酸和香草酸),按“2.1.2.1”项下均制备6份供试品并进样分析,记录峰面积,计算各成分含量及rsd值。
[0164]
结果见表4,表明测定方法重复性好。
[0165]
表4、重复性考察
[0166][0167]
(5)稳定性试验
[0168]
取根皮酚-β-d-葡糖苷标准品(7.545μg
·
ml-1
)、5-羟甲基-2-呋喃甲酸标准品(1.265μg
·
ml-1
)、香草酸标准品(1.315μg
·
ml-1
)混合标准品,按“2.1.2.1”项下条件进样分析,在0、2、4、8、12h分别进样测定,记录峰面积,计算rsd值。取陈皮样品s2(根皮酚-β-d-葡糖苷)及s4(5-羟甲基-2-呋喃甲酸和香草酸),精密称定,按“2.1.2.1”项下制备供试品并进样分析,在0、2、4、8、12h分别进样测定,记录峰面积,计算rsd值。
[0169]
结果见表5。结果表明,在12h内,三种标准品计算rsd均《2%,陈皮样品供试品计算rsd值均《1%。表明12h内对照品和供试品稳定性良好。
[0170]
表5、稳定性考察
[0171][0172]
(6)加样回收试验
[0173]
分别取s2陈皮样品0.25g,精密称定,以当前样品含量的1:1的比例加入根皮酚-β-d-葡糖苷标准品,取s4陈皮样品0.25g,精密称定,以当前样品含量的1:1的比例加入根皮酚-β-d-葡糖苷标准品,全部样品均按“2.1.2.1”项下制备供试品并进样分析,记录峰面积,计算含量,每个重复3次。计算平均回收率及rsd。
[0174]
根据2020版《中国药典》中规定,陈皮样品中根皮酚-β-d-葡糖苷的含量属于1000ppm级别,加样回收率应在90%~108%,5-羟甲基-2-呋喃与香草酸的含量属于100ppm级别,加样回收率应在85%~110%,如表6所示,陈皮样品中根皮酚-β-d-葡糖苷平均加样回收率为94.33%,rsd为0.43%;5-羟甲基-2-呋喃甲酸平均加样回收率为105.18%,rsd为0.28%;香草酸平均加样回收率为103.8%,rsd为0.19%,三种成分的加样回收率均符合标准。
[0175]
表6、加样回收考察
[0176][0177]
2.1.2.3含量测定结果
[0178]
陈皮样品精密称取0.5g,每个样品平行制作2份,按“2.1.2.1”项下制备供试品并进样分析,计算并记录三个成分的含量,结果见表7。
[0179]
表7、样品中三个成分的含量(n=2)
[0180][0181]
2.1.3结果与分析
[0182]
由表7可知,以上三种成分含量在不同存储容器、不同年份的陈皮样品中均存在较大差异。其中,成分“根皮酚-β-d-葡糖苷”含量在s2~s4三个样品中呈现的趋势为s2(1.514)>s3(0.301)>s4(0.227),结合表1、表2可知,三个样品存储容器的透气性良好程度依次为s2(mf)<s3(ck)<s4(dz),其储藏年份、来源等其他条件均一致;由此可见,陈皮在陈化过程中存储容器的透气性越良好,其“根皮酚-β-d-葡糖苷”含量就越少。在相同存储容器、不同年份陈皮中,成分“根皮酚-β-d-葡糖苷”的含量也呈现出“年份越高,含量越少”的变化趋势,如s2(1.514)<s1(3.458)、s5(0.209)<s3(0.301)。该结果表明,“根皮酚-β-d-葡糖苷”是在陈化过程中逐渐“消失”(简称“消”)的成分。
[0183]
然而,对于成分“香草酸”而言,其含量在不同存储容器、相同年份陈皮中的大小依次为s2(0.042)<s3(0.144)<s4(0.269),呈现出“存储容器透气性越好,则含量越高”的变化趋势;在相同存储容器、不同年份陈皮中的大小依次为s1(0.025)<s2(0.042)、s3(0.144)<s5(0.243)、s4(0.269)<s6(0.333),均呈现出“年份越高,则含量越高”的变化趋势。该结果表明,“香草酸”是在陈化过程中逐渐“增长”(简称“长”)的成分。
[0184]
对于成分“5-羟甲基-2-呋喃甲酸”而言,其含量在不同存储容器、相同年份陈皮
中,与成分“香草酸”的变化趋势一致,即“存储容器透气性越好,则含量越高”。然而,在相同存储容器、不同年份陈皮中出现了不一致的变化趋势,如s1(0.007)>s2(0.006)、s3(0.153)>s5(0.133)、s4(0.335)<s6(0.389);但“5-羟甲基-2-呋喃甲酸”含量在总体上表现出“年份越高,则含量越高”的趋势。以上结果表明,也可以认为“5-羟甲基-2-呋喃甲酸”是在陈化过程中逐渐“增长”(简称“长”)的成分。
[0185]
为了更加直观地体现陈皮在陈化过程中存在消长变化的差异成分含量与陈化年份之间的关联性,可将逐渐“长”的成分与“消”成分二类成分合二为一,即比值当中“长”的成分作为分子,“消”的成分作为分母,比值结果见表8。
[0186]
表8、“消”、“长”变化成分之间的含量比值
[0187][0188]
由表8可知,比值“香草酸/根皮酚-β-d-葡糖苷”在不同存储容器、相同年份的陈皮样品中,比值大小依次为s4(1.186)>s3(0.478)>s2(0.028),呈现出“存储容器透气性越好,则含量越高”的变化趋势;在不同年份、相同存储容器的陈皮样品中,比值大小依次为s1(0.007)<s2(0.028)、s3(0.478)<s5(1.162)、s4(1.185)<s6(1.313),均呈现“年份高的,比值越大”的变化趋势。比值“5-羟甲基-2-呋喃甲酸/根皮酚-β-d-葡糖苷”在不同存储容器、相同年份的陈皮样品中的变化趋势,与比值“香草酸/根皮酚-β-d-葡糖苷”的一致。结果表明,比值“香草酸/根皮酚-β-d-葡糖苷”、“5-羟甲基-2-呋喃甲酸/根皮酚-β-d-葡糖苷”与陈化年份均存在较强的关联性。
[0189]
同时,比较不同存储容器(dz与ck)、年份相差较大(2017与2007)的2个陈皮样品s4、s5比值可知,2个比值大小均为s4(1.185)>s5(1.162)、s4(1.476)>s5(0.637);该结果表明,存储容器对比值的影响大于年份对比值的影响。
[0190]
2.2陈皮陈化过程中挥发油类成分之间的比值测定
[0191]
陈皮在陈化过程中气味也会发生明显的变化,低年份的陈皮,闻起来为果香味、清香味;高年份的陈皮,则闻起来为药香陈味,陈香醇厚。而关于广陈皮挥发油的相关研究与标准已有相关报道,其中地方标准《db44t604-2009地理标志产品新会陈皮》就规定“新会陈皮的气相色谱图谱中至少出现三个强峰,即柠檬烯、γ-松油烯(又称“γ-萜品烯”)、2-(甲氨基)苯甲酸甲酯”,因此本研究以该三种挥发油成分作为目标成分进行测定,计算各成分相对百分含量之间的比值,并分析比值与陈皮年份之间的相关性;最后,将与陈化年份相关性强的比值纳入陈化度的量化公式中。
[0192]
2.2.1色谱条件
[0193]
采用安捷伦hp-5ms(30mm
×
0.25mm
×
0.25μm)色谱柱,高纯he作为载气,流速为1.0ml/min,进样口温度为230℃,采用程序升温模式,柱初始温度为50℃,保持2min;以3℃/
min的速率升至70℃,保持10min;以8℃/min的速率升至110℃,保持5min;以4℃/min的速率升至210℃,保持2min。分流比为50:1。
[0194]
质谱(ms)条件:离子源温度230℃,电子能量70ev,离子采集范围35-450m/z。陈皮样品gc/ms色谱图,见图18。
[0195]
2.2.2样品前处理方法
[0196]
将陈皮样品剪碎边长1cm的小方块,取0.5g于40ml spme专用采样瓶中,密封后于90℃平衡10min,插入固相萃取纤维50/30μm car/dvb/pdms顶空萃取40min,250℃解吸3min进样。
[0197]
2.2.3gc/ms测定
[0198]
采用顶空固相微萃取(hs-spme)与气相色谱-质谱联用(gc/ms)技术测定所有陈皮样品,每组取样设置2组平行实验,所得质谱数据与nist14数据库进行匹配,以确定目标成分的真实性。
[0199]
2.2.4结果与分析
[0200]
陈皮样品中“柠檬烯”、“γ-萜品烯”、“2-(甲氨基)苯甲酸甲酯”三种挥发油成分相对百分含量的测定结果,见表9。
[0201]
表9、三种挥发油类成分的相对含量及其比值结果(n=2)
[0202][0203]
由表9可知,以上三种挥发油成分相对含量在不同存储容器、不同年份的陈皮样品中均存在较大差异。由于检测各样品挥发油成分时,色谱响应峰数量的各不相同,即基数不一致,导致所测得的单个挥发油相对含量数据之间无法进行比较。为了避免此问题对数据分析产生的影响,本研究采用挥发油成分相对含量之间的比值作为分析数据,两两成分之间的比值结果见表9。
[0204]
2.3陈皮陈化度的构建及其评价方法
[0205]
由表8可知,通过比较相同存储容器(均为ck)、年份相差较大(2017与2007)的2个陈皮样品比值大小,可以得知样品s3、s5的比值“香草酸/根皮酚-β-d-葡糖苷”分别为0.478、1.162,两者大小相差近三倍,该数据差距与其10年的年份差距较匹配;而比值“5-羟甲基-2-呋喃甲酸/根皮酚-β-d-葡糖苷”分别为0.508、0.637,两者大小相当,该数据差距与其10年的年份差距不匹配。同时,鉴于经过高温加速处理的陈皮会产生成分“5-羟甲基糠醛”,而该成分在特定条件下能转化为“5-羟甲基-2-呋喃甲酸”。因此,比值“香草酸/根皮酚-β-d-葡糖苷”较“5-羟甲基-2-呋喃甲酸/根皮酚-β-d-葡糖苷”更加适合作为陈皮陈化度的评价指标。
[0206]
同时,分析表8中相同存储容器、不同年份陈皮中的比值“香草酸/根皮酚-β-d-葡
糖苷”大小,可知存储容器透气性良好的四年以上陈皮样品(s4,s6)比值均>0.5,透气性较差的四年以下陈皮样品(s1、s2)比值均<0.5;存储容器透气性一般的七年陈皮样品s5比值(1.162)大于0.5,而存储容器透气性一般的四年陈皮样品s3比值(0.479)也小于0.5。根据上述得到的结论“存储容器对比值的影响大于年份的”可以推测,存储容器透气性一般(ck)的四年陈皮的陈化年份小于存储容器透气性良好(dz)的三年陈皮的陈化年份;再结合地方标准《db44/t 604-2009地理标志产品新会陈皮》规定陈皮需陈化三年及以上;据此可以认为样品s1、s2、s3均未得到有效的陈化。因此,可将比值“香草酸/根皮酚-β-d-葡糖苷”是否大于0.5,作为判断陈皮是否得到有效陈化的重要依据。
[0207]
从表9中可以得知,对于相同年份、不同存储容器的陈皮样品(s2~s4)而言,其比值“柠檬烯/γ-萜品烯”大小依次为s2(4.60)<s3(5.20)<s4(5.27),呈现出“存储容器透气性越好,则比值越大”的变化趋势;在不同存储容器、相同年份陈皮中,该比值大小也依次为s1(3.47)<s2(4.60)、s3(5.20)<s5(7.03)、s4(5.27)<s6(14.39),均呈现出“年份越高,则比值越大”的变化趋势。由此可见,陈皮在陈化过程中比值“柠檬烯/γ-萜品烯”呈不断增加的趋势,说明陈化年份与比值“柠檬烯/γ-萜品烯”之间存在较强的关联性。而比值“2-(甲氨基)苯甲酸甲酯/柠檬烯”、“2-(甲氨基)苯甲酸甲酯/γ-萜品烯”却不存在此类变化趋势。因此,可以将比值“柠檬烯/γ-萜品烯”作为陈皮陈化度的评价指标。
[0208]
综上所述,通过整合陈皮中比值“香草酸/根皮酚-β-d-葡糖苷”与“柠檬烯/γ-萜品烯”,并结合陈皮是否得到有效的陈化等因素,最终构建出陈皮陈化度(chd)的量化公式:
[0209][0210]
注:式中d1为酸类成分香草酸含量;d2为糖苷类成分根皮酚-β-d-葡糖苷含量;h1为挥发油类成分柠檬烯相对含量;h2为挥发油类成分γ-萜烯品相对含量;
[0211]
陈皮的chd值越大,说明陈皮的陈化年份越高,其品质越优良。其中,若chd值<0,说明陈皮的未能得到有效的陈化,即陈化年份低。
[0212]
根据上述陈皮陈化度的量化公式,可知试验样品s1~s6的chd值大小分别为-1.74、-2.17、-0.11、3.62、4.67、11.93;再结合表1、表2、表8、表9中的信息与数据,构建出试验样品陈化度的变化趋势图,见图19。
[0213]
3陈化度的应用
[0214]
3.1验证样品信息
[0215]
为验证陈化度量化方法的可行性,选择不同存储容器、不同年份的陈皮作为验证样品,样品信息详见表10。
[0216]
表10、验证样品信息
[0217][0218]
注:“——”代表信息不详
[0219]
3.2验证样品中成分含量比值及陈化度的测定
[0220]
验证样品中糖苷、酸类成分的色谱条件与供试液制备方法,按“2.1.2.1”项下方法进行测定与制备;挥发油类成分的色谱条件与样品前处理方法,则分别按“2.2.1”、“2.2.2”项下方法进行测定与处理,其含量比值结果见表11。
[0221]
根据“2.3”项下陈皮陈化度的量化公式,计算样品的陈化度(chd),结果见表11;再结合表10中的样品信息,构建出验证样品的陈化度变化趋势图,见图20。
[0222]
表11、验证样品中成分含量比值及陈化度
[0223][0224]
由表11、图20可知,验证样品中chd值的变化趋势与试验样品的变化趋势(图19)一致,即陈皮在陈化过程中的陈化度呈不断上升的变化规律。
[0225]
结合图19与图20中的数据可知,存储容器为mf的2020年、2017年、2015年产s1、s2、s8陈皮的chd值(分别为-1.74、-2.17、-2.16)均小于0,该结果表明,用透气性差的存储容器储藏陈皮,无论其储藏多久,都达不到有效的陈化。
[0226]
存储容器为ck的2017年、2016年、2007年产s3、s7、s5陈皮的chd值大小分别为-0.11、0.58、4.67,该结果显示,用透气性一般的存储容器储藏陈皮,储藏到一定的年份(五年及其以上),其chd值大小也能大于0,即得到有效的陈化;储藏时间不够(低于五年)的ck陈皮,则不能得到有效的陈化。
[0227]
存储容器为dz的2017年、2014年、2014年、2005年产s4、s9、s10、s6陈皮的chd值大小分别为3.62、5.04、6.46、11.93,该结果表明,用透气性良好的存储容器储藏陈皮,在较短的储藏时间内就能得到有效的陈化。同时,比较存储容器透气性与生产年份相反的三个陈皮样品s4(dz、2017年)、s7(ck、2016年)及s8(mf、2015年)陈化度大小,其chd值表现为s4(3.62)>s7(0.58)>s8(-2.16),陈皮时间越久,其chd值反而越小;该结果表明,存储容器对陈化度的影响大于储藏年份对陈化度的影响。因此,根据上述三个存储容器透气性与生产年份相反的陈皮样品chd值,可以推测存储容器为dz的三年陈皮chd值应该也大于0。该结论科学诠释了地方标准《db44/t 604-2009地理标志产品新会陈皮》中“陈皮需陈化三年及以上”的规定。
[0228]
3.3结论
[0229]
关于陈皮陈化度公式中所涉及含量测定指标成分与挥发油成分。其中,对于挥发油成分“柠檬烯”、“γ-萜品烯”而言,其属于地方标准《db44t604-2009地理标志产品新会陈皮》所规定的“新会陈皮的气相色谱图谱中至少出现三个强峰,即柠檬烯、γ-松油烯(又称“γ-萜品烯”)、2-(甲氨基)苯甲酸甲酯”三个挥发油成分中的二个。对于含量测定指标成分“根皮酚-β-d-葡糖苷”、“香草酸”而言,相关研究表明,香草酸具有抗氧化、抗炎等药理作用,根皮酚-β-d-葡糖苷具有降低血糖、改善记忆力、抗氧化、抗癌等多种重要的生物活性。
综上所述,陈皮陈化度公式中的四个指标成分均属于陈皮的有效成分,均可作为评价陈皮品质的指标。
[0230]
因此,通过陈皮的chd值大小,判断陈皮是否得到有效的陈化以及陈化年份的高低是可行的。
[0231]
陈皮陈化度(chd)的量化公式:
[0232][0233]
注:式中d1为酸类成分香草酸含量;d2为糖苷类成分根皮酚-β-d-葡糖苷含量;h1为挥发油类成分柠檬烯相对含量;h2为挥发油类成分γ-萜烯品相对含量。
[0234]
陈皮的chd值越大,说明陈皮的陈化年份越高,其品质越优良。其中,若chd值<0,说明陈皮的未能得到有效的陈化,即陈化年份低。
[0235]
综上,本发明的上述方法:一方面,采用uplc-pad技术,找出陈皮陈化过程中存在变化规律的差异成分,并结合制备型hplc系统、核磁共振技术进行分离、鉴定;然后采用uplc技术测定“消”、“长”变化成分的含量;以“长”的成分含量作为分子,“消”的成分含量作为分母,计算两类成分含量之间的比值。另一方面,采用固相微萃取-气相色谱-质谱联用技术(spme-gc-ms)等技术组合,定性半定量分析挥发油类成分,并计算挥发油成分相对含量之间的比值。接着,分析上述成分含量比值与陈化年份之间的关联性,找出与陈化年份关联性较强的比值,并将该比值作为陈化度公式中的变量。本发明的技术路线图见图21。
[0236]
以上所述实施方式和实施例的各技术特征可以进行任意合适方式的组合,为使描述简洁,未对上述实施方式和实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为在本说明书记载的范围中。
[0237]
以上所述实施例仅表达了本发明的几种实施方式,便于具体和详细地理解本发明的技术方案,但并不能因此而理解为对发明专利保护范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,得到的等价形式同样落于本技术的保护范围。还应当理解,本领域技术人员在本发明提供的技术方案的基础上,通过合乎逻辑的分析、推理或者有限的试验得到的技术方案,均在本发明所附权利要求的保护范围内。因此,本发明专利的保护范围应以所附权利要求的内容为准,说明书及附图可以用于解释权利要求的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1