二苯乙烯类化合物在制备抗肿瘤药物中的用途的制作方法
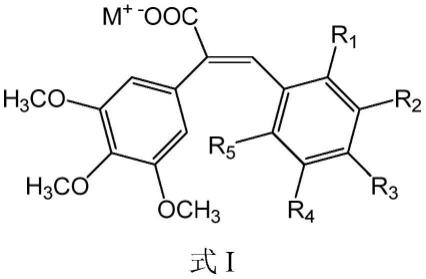
1.本发明涉及药物领域,具体涉及二苯乙烯类化合物在制备抗肿瘤药物中的用途。
背景技术:
2.cd8
+
t淋巴细胞是一种细胞毒性的t淋巴细胞,能够杀灭肿瘤细胞,阻断肿瘤扩散。如果药物使用后如果能显著提高实体瘤组织中的cd8+t淋巴细胞的细胞浓度,使cd8+t淋巴细胞富集在实体瘤组织中,可实现药物抗癌作用。
3.此外,恶性肿瘤的内部供血和恶性肿瘤周边的血管都是很丰富的,一种药物如果能作用于肿瘤血管内皮细胞,使得活跃的肿瘤血管出现炎症反应并肿胀而阻塞血管,切断血液在肿瘤中的流动,则可以使肿瘤细胞“断养”后逐渐饥饿死亡,实现抗肿瘤的作用。
4.发明人前期研究发现苯乙烯酸衍生物具有一定的肿瘤血管的特异性靶向性作用,可以抑制肿瘤生长,但不能杀灭肿瘤细胞,特别是实体瘤组织边缘部分的肿瘤细胞,从而引起肿瘤组织通过边缘部分的快速增殖,治疗肿瘤效果不佳。
5.如果能够提供一种既能使cd8
+
t淋巴细胞富集在实体瘤组织中,又能够阻止血液在肿瘤中的流动的药物,将在抗肿瘤的应用中具有很大的应用前景。
技术实现要素:
6.本发明的目的是提供二苯乙烯类化合物在制备抗肿瘤药物中的用途。
7.本发明提供了式i所示化合物、或其盐、或其立体异构体、或其溶剂合物在制备抗肿瘤药物中的用途:
[0008][0009]
其中,m选自氢、钠、钾、钙、铵或有机胺;
[0010]
r1、r2、r3、r4、r5分别独立选自氢、c1~c6烷基、c1~c6烷氧基、羟基、氨基、卤素。
[0011]
进一步地,所述有机胺为氨基酸、肽、叔丁胺或正丁胺;
[0012]
和/或,r1、r2、r3、r4、r5分别独立选自氢、c1~c3烷基、甲氧基、乙氧基、羟基、氨基、氟、氯或溴;
[0013]
优选地,
[0014]
所述有机胺选自葡甲胺、吡嗪、丙氨酸、正丁胺、赖氨酸、叔丁胺;
[0015]
和/或,r1、r2、r3、r4、r5分别独立选自氢、甲氧基、乙氧基、羟基、氟、氯或溴。
[0016]
进一步地,所述化合物为式ii所示:
[0017][0018]
其中,m选自氢、钠、钾、钙、铵、葡甲胺、吡嗪、丙氨酸、正丁胺、赖氨酸或叔丁胺;
[0019]
r1、r2、r3、r4分别独立选自氢、甲氧基、乙氧基、羟基、氟、氯或溴。
[0020]
进一步地,所述化合物为式iii所示:
[0021][0022]
其中,m选自氢、钠、钾、钙、铵、葡甲胺、吡嗪、丙氨酸、正丁胺、赖氨酸或叔丁胺;
[0023]
r1、r2、r3分别独立选自氢、甲氧基、乙氧基、羟基、氟、氯或溴。
[0024]
进一步地,所述化合物为式iv所示:
[0025][0026]
其中,m选自氢、钠、钾、钙、铵、葡甲胺、吡嗪、丙氨酸、正丁胺、赖氨酸或叔丁胺;
[0027]
r2、r3分别独立选自氢、甲氧基、乙氧基、羟基、氟、氯或溴。
[0028]
进一步地,所述药物为具有富集实体瘤内cd8
+
t淋巴细胞和/或破坏肿瘤血管内壁功效的药物;
[0029]
优选地,所述药物为引起实体瘤中心部位肿瘤细胞坏死的药物。
[0030]
进一步地,所述药物为预防和/或治疗肺癌、肝癌、胃癌、卵巢癌、前列腺癌或肾细胞癌的药物。
[0031]
进一步地,所述药物为预防和/或治疗肺癌、肝癌、胃癌或卵巢癌的药物时,式i所
示化合物的结构为式va所示:
[0032][0033]
进一步地,所述药物为预防和/或治疗前列腺癌或肾细胞癌的药物时,式i所示化合物的结构为式vb所示:
[0034][0035]
本发明还提供了一种治疗肿瘤的药物制剂,它是由前述的化合物、或其盐、或其立体异构体、或其溶剂合物为活性成分,加上药学上可接受的辅料或辅助性成分制备而成的药物制剂;
[0036]
优选地,所述药物制剂为注射制剂、口服制剂或外用制剂。
[0037]
本发明还提供了前述的化合物、或其盐、或其立体异构体、或其溶剂合物和肿瘤免疫治疗药物联用在制备抗肿瘤联合用药物中的用途;
[0038]
优选地,所述肿瘤免疫治疗药物为pd1、pd-l1、t细胞、nk细胞或cart细胞。
[0039]
进一步地,式vc所示化合物、或其盐、或其立体异构体、或其溶剂合物和pd-l1联用在制备预防和/或治疗肺癌的联合用药物中的用途;
[0040][0041]
进一步地,式va所示化合物、或其盐、或其立体异构体、或其溶剂合物和t细胞联用
在制备预防和/或治疗肝癌的联合用药物中的用途;
[0042][0043]
本发明的发明人之前研究的苯乙烯酸衍生物虽然具有一定的肿瘤血管的特异性靶向性作用,但只能破坏肿瘤血管,抑制肿瘤生长,不能杀灭肿瘤细胞,特别是实体瘤组织边缘部分的肿瘤细胞,从而引起肿瘤组织通过边缘部分的快速增殖。在前期工作的基础上,发明人通过结构优化及体内筛选,研究出本发明系列化合物具有富集实体瘤内t淋巴细胞cd8+及破坏新生血管内皮细胞黏附蛋白双重作用,可用于制备抗肿瘤药物。
[0044]
以式(i)结构的二苯乙烯类化合物为有效活性成分,与药物中可以接受的相应药用辅料或载体等辅助添加成分组合,并按相应的常规制药方法加工处理后,可以制备成为口服、注射或外用型等相应形式的血管靶向剂药物。例如,与口服药物制剂中可以被接受的崩解剂、赋形剂、润滑剂、粘合剂、填充剂等常用的辅助添加成份混合后,按相应的常规工艺方法处理,即可制成为片剂、丸剂、颗粒剂、胶囊剂或适当形式的缓释剂、控释剂等固体制剂形式的口服药物;按注射药物制剂中允许使用的适当溶剂和附加剂配合及相应的工艺操作处理后,可以制备成相应的水针或粉针等肌肉或静脉形式的注射制剂药物;与常用的溶剂、稳定激或聚丙烯酸钠、聚乙烯醇、聚乙二醇、甘油、植物油、凡士林、羊毛脂等亲油性基质和/或水溶性基质混合,按相应的外用药物制剂处理,即可制成为滴眼液或贴膏剂、膏药、凝胶剂、软膏剂、搽剂、涂膜剂等。这些注射液、无菌粉针、无菌冻干粉针等注射制剂,片剂、胶囊、各种缓控释制剂等口服制剂,以及如滴眼液、软膏等外用制剂形式药物等,可分别供治疗抗实体瘤药物使用。
[0045]
本发明提供了二苯乙烯类化合物在制备抗肿瘤药物中的用途,本发明研究发现部分二苯乙烯类化合物具有富集实体瘤内t淋巴细胞cd8
+
及靶向破坏肿瘤血管内壁的双重作用,在通过杀伤性t淋巴细胞抗原cd8
+
消灭肿瘤细胞的同时,切断肿瘤组织的血供,造成实体瘤内部的快速坏死,大大提高对肿瘤的杀伤力。本发明二苯乙烯类化合物可用于制备抗肿瘤药物,也可以与肿瘤免疫治疗药物联合使用,发挥协同增效抗肿瘤功效。这些二苯乙烯类化合物中不同结构的化合物抗肿瘤功效不同,可用于制备不同的抗肿瘤药物,具有良好的应用前景。
[0046]
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
[0047]
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
[0048]
图1为定量蛋白质组实验流程图。
[0049]
图2为人体肺癌95d的he染色结果。
[0050]
图3为人体肝癌be17404的he染色结果。
[0051]
图4为人体肺癌95d的pcna免疫组化染色结果。
[0052]
图5为人体肝癌be17404的pcna免疫组化染色结果。
具体实施方式
[0053]
本发明具体实施方式中使用的原料、设备均为已知产品,通过购买市售产品获得。
[0054]
本发明二苯乙烯类化合物的结构如式(i)所示:
[0055][0056]
式中,m选自氢、钠、钾、钙、铵或包括氨基酸、叔丁胺、正丁胺、肽在内的有机胺;r1、r2、r3、r4、r5分别选自氢、烷基、羟基、甲氧基、乙氧基、胺基、氟、氯或溴。
[0057]
本发明具体化合物如表1所示:
[0058]
表1.本发明化合物具体结构
[0059]
化合物r1r2r3r4r5m1羟基羟基甲氧基氢氢氢2羟基羟基甲氧基氢氢葡甲胺3氢甲氧基氢甲氧基氢钠4氢氟甲氧基氢氢吡嗪5氢羟基甲氧基氯氢丙氨酸6氢羟基甲氧基氢氢正丁胺7氢甲氧基羟基氢氢赖氨酸8氢羟基甲氧基氟氢叔丁胺9氟羟基甲氧基氢氢正丁胺10氢甲氧基氟甲氧基氢钾11氢羟基甲氧基羟基氢正丁胺12甲氧基羟基甲氧基氢氢赖氨酸13氢羟基乙氧基氢氢赖氨酸
[0060]
实施例1、本发明化合物1的制备
[0061][0062]
制备方法为:在50ml三口烧瓶中加入2.26g(0.010mol)3,4,5-三甲氧基苯乙酸、2.02g(0.012mol)2,3-二羟基-4-甲氧基苯甲醛,再加入3.2ml醋酸酐和1.6ml三乙胺,摇匀,装入磁力搅拌子,装上回流冷凝管,通入氮气保护,油浴加热升温到150℃,保温反应5小时,冷却至80℃,将回流冷凝管换为滴液漏斗,滴入8ml盐酸溶液,保持80℃反应1小时,冷却至室温,滴入40%氢氧化钠溶液2ml,继续反应1小时,然后滴入1.8ml浓盐酸,析出固体之后继续搅拌反应1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体(化合物1)2.17g(收率57.7%)。1h nmr(400mhz,dmso)3.672(d 9h 3’,4
′
,5
’‑
och3),3.821(d 3h 4
”‑
och3),5.265(s 1h 2
”‑
oh),5.318(s 1h 3
”‑
oh),6.273(s 1h 2
’‑
h),6.348(d 1h 6
’‑
h),6.386(d 1h 5
”‑
h),7.415(s 1h 3-h);10.982(s 1h co-oh);
13
c nmr(100mhz,dmso)δ57.692,57.826,62.837,109.313,112.713,118.627,125.392,129.883,137.423,138.153,138.232,138.772,146.391,149.167,154.482,178.277共16组碳。
[0063]
实施例2、本发明化合物2的制备
[0064][0065]
制备方法为:在10ml三口烧瓶中加入1.82g(4.84mmol)(e)-3-(2,3-二羟基-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸(化合物1)、0.99g(5.08mmol)葡甲胺,再加入15ml乙醇,摇匀,装入磁力搅拌子,水浴加热升温到60℃,溶清后冷却至室温,析出固体后,继续搅拌1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体(化合物2)2.21g(收率80.0%)。1h nmr(400mhz,dmso)2.791(d,3h a-ch3),3.187(d,2h a-ch2),3.297(d,1h a-ch),3.365(d,1h a-ch),3.382(d,1h a-ch),3.516(d,2h a-ch2),3.683(d,4h a-oh),3.698(d,1h a-oh),3.713(d 9h 3’,4’,5
’‑
och3),3.796(d 3h 4
”‑
och3),5.159(s 1h 2
”‑
oh),5.435(s 1h 3
”‑
oh),6.368(s 1h 2
’‑
h),6.521(d 1h 6
’‑
h),6.472(d 1h 5
”‑
h),7.477(s 1h 3-h);
13
c nmr(100mhz,dmso)δ34.286,51.217,57.692,
57.826,62.837,63.295,68.786,71.198,73.136,73.982,109.313,112.713,118.627,125.392,129.883,137.423,138.153,138.232,138.772,146.391,149.167,154.482,178.277共23组碳。
[0066]
实施例3、本发明化合物3的制备
[0067][0068]
制备方法为:在50ml三口烧瓶中加入2.26g(0.010mol)3,4,5-三甲氧基苯乙酸、1.99g(0.012mol)3,5-二甲氧基苯甲醛,再加入3.2ml醋酸酐和1.6ml三乙胺,摇匀,装入磁力搅拌子,装上回流冷凝管,通入氮气保护,油浴加热升温到150℃,保温反应5小时,冷却至80℃,将回流冷凝管换为滴液漏斗,滴入8ml盐酸溶液,保持80℃反应1小时,冷却至室温,滴入40%氢氧化钠溶液2ml,继续反应1小时,然后滴入1.8ml浓盐酸,析出固体之后继续搅拌反应1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体2.25g(收率60.1%)。类白色结晶性固体为(e)-3-(3,5-二甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸。
[0069]
在10ml三口烧瓶中加入1.81g(4.84mmol)(e)-3-(3,5-二甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸、10ml无水乙醇,40%氢氧化钠溶液0.5ml,摇匀,装入磁力搅拌子,搅拌,析出固体后,继续搅拌1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体(化合物3)1.62g(收率84.4%)。1h nmr(400mhz,dmso)3.727(d 9h 3’,4’,5
’‑
och3),3.869(d 6h 3”,5
”‑
och3),6.174(d 1h 4
”‑
h),6.273(s 1h 2
’‑
h),6.348(d 1h 6
’‑
h),6.386(d 1h 5
”‑
h),6.723(d 2h 2”,5
”‑
h)),7.329(s 1h 3-h);
13
c nmr(100mhz,dmso)δ55.823,57.314,62.729,108.476,113.626,117.196,126.181,128.347,136.973,137.826,137.951,137.865,147.127,148.331,154.672,171.323共16组碳。
[0070]
实施例4、本发明化合物4的制备
[0071][0072]
制备方法为:在50ml三口烧瓶中加入2.26g(0.010mol)3,4,5-三甲氧基苯乙酸、1.85g(0.012mol)3-氟-4-甲氧基苯甲醛,再加入3.2ml醋酸酐和1.6ml三乙胺,摇匀,装入磁力搅拌子,装上回流冷凝管,通入氮气保护,油浴加热升温到150℃,保温反应5小时,冷却至80℃,将回流冷凝管换为滴液漏斗,滴入8ml盐酸溶液(1+5),保持80℃反应1小时,冷却至室温,滴入40%氢氧化钠溶液2ml,继续反应1小时,然后滴入1.8ml浓盐酸,析出固体之后继续搅拌反应1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体2.15g(收率59.3%)。类白色结晶性固体为(e)-3-(3-氟-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸。
[0073]
在10ml三口烧瓶中加入1.75g(4.84mmol)(e)-3-(3-氟-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸、10ml无水乙醇,0.41g吡嗪,摇匀,装入磁力搅拌子,搅拌,析出固体后,继续搅拌1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体(化合物4)1.76g(收率82.2%)。1h nmr(400mhz,dmso)δ3.612(d 9h 3’,4’,5
’‑
och3),3.824(d 3h 4
”‑
och3),6.287(s 2h 2’,6
’‑
h),6.413(s 1h 2
”‑
h),6.578(d 1h 6
”‑
h),6.612(d 1h 5
”‑
h),9.583(s 2h c-h),9.689(s 2h c-h),9.823(s 1h 3-h);
13
c nmr(100mhz,dmso)δ56.276,57.327,63.196,108.893,113.746,117.845,125.637,130.762,138.524,138.267,139.241,139.482,142.158,146.783,148.298,152.930,174.128共17组碳。
[0074]
实施例5、本发明化合物5的制备
[0075][0076]
制备方法为:在50ml三口烧瓶中加入2.26g(0.010mol)3,4,5-三甲氧基苯乙酸、
2.24g(0.012mol)3-氯-5-羟基-4-甲氧基苯甲醛,再加入3.2ml醋酸酐和1.6ml三乙胺,摇匀,装入磁力搅拌子,装上回流冷凝管,通入氮气保护,油浴加热升温到150℃,保温反应5小时,冷却至80℃,将回流冷凝管换为滴液漏斗,滴入8ml盐酸溶液,保持80℃反应1小时,冷却至室温,滴入40%氢氧化钠溶液2ml,继续反应1小时,然后滴入1.8ml浓盐酸,析出固体之后继续搅拌反应1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体2.13g(收率53.9%)。类白色结晶性固体为(e)-3-(3-氯-5-羟基-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸。
[0077]
在10ml三口烧瓶中加入1.75g(4.84mmol)(e)-3-(3-氯-5-羟基-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸、0.45g(5.08mmol)丙氨酸,10ml无水乙醇,摇匀,装入磁力搅拌子,搅拌,析出固体后,继续搅拌1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体(化合物5)1.42g(收率60.6%)。[m+h]
+
:443.16。1h-nmr(cdcl3,δ(ppm)):9.972(2h),9.712(2h),7.326(1h),6.887(1h),6.865(1h),6.853(1h),3.812(3h),3.924(9h),6.615(2h)。
[0078]
实施例6、本发明化合物6的制备
[0079][0080]
制备方法为:在50ml三口烧瓶中加入2.26g(0.010mol)3,4,5-三甲氧基苯乙酸、1.83g(0.012mol)异香草醛,再加入3.2ml醋酸酐和1.6ml三乙胺,摇匀,装入磁力搅拌子,装上回流冷凝管,通入氮气保护,油浴加热升温到150℃,保温反应5小时,冷却至80℃,将回流冷凝管换为滴液漏斗,滴入8ml盐酸溶液,保持80℃反应1小时,冷却至室温,滴入40%氢氧化钠溶液12ml,继续反应1小时,然后滴入10ml浓盐酸,析出固体之后继续搅拌反应1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体2.56g(收率71.0%)。类白色结晶性固体为(e)-3-(3-羟基-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸。
[0081]
在10ml三口烧瓶中加入1.74g(4.84mmol)(e)-3-(3-羟基-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸、0.37g(5.08mmol)正丁胺,10ml无水乙醇,摇匀,装入磁力搅拌子,搅拌,析出固体后,继续搅拌1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体(化合物6)1.68g(收率80.1%)。1h nmr(400mhz,dmso)δ0.832(d,3h a-ch3),1.329(d 2h b-ch3),1.546(d 2h c-ch2),2.914(d 2h,d-ch3),3.593(d 9h 3’,4’,5
′‑
och3),3.749(d 3h 4
”‑
och3),6.375(s 2h 2
′‑
h),6.386(s 1h 2
”‑
h),6.462(d 1h 6
”‑
h),6.499(d 1h 5
”‑
h),7.346(s 1h 3-h);
13
c nmr(100mhz,dmso)δ15.103,21.343,31.101,41.526,57.788,58.186,63.341,109.224,113.626,118.900,125.518,130.918,137.718,137.812,138.175,139.291,146.588,149.858,154.930,178.099共20组碳。
[0082]
实施例7、本发明化合物7的制备
[0083][0084]
制备方法为:在50ml三口烧瓶中加入2.26g(0.010mol)3,4,5-三甲氧基苯乙酸、1.83g(0.012mol)香草醛,再加入3.2ml醋酸酐和1.6ml三乙胺,摇匀,装入磁力搅拌子,装上回流冷凝管,通入氮气保护,油浴加热升温到150℃,保温反应5小时,冷却至80℃,将回流冷凝管换为滴液漏斗,滴入8ml盐酸溶液,保持80℃反应1小时,冷却至室温,滴入40%氢氧化钠溶液2ml,继续反应1小时,然后滴入1.8ml浓盐酸,析出固体之后继续搅拌反应1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体2.74g(收率76.0%)。类白色结晶性固体为(e)-3-(4-羟基-3-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸。
[0085]
在10ml三口烧瓶中加入1.74g(4.84mmol)(e)-3-(4-羟基-3-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸、0.74g(5.08mmol)赖氨酸,再加入15ml乙醇,摇匀,装入磁力搅拌子,水浴加热升温到60℃,溶清后冷却至室温,析出固体后,继续搅拌1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体(化合物7)1.75g(收率71.4%)。[m+h]
+
:507.23。1h-nmr(cdcl3,δ(ppm)):1.187(2h),1.742(2h),2.116(2h),3.281(2h),3.373(1h),5.095(2h),7.386(1h),3.659(3h),5.287(1h),7.137(1h),7.349(1h),3.876(9h),6.517(2h)。
[0086]
实施例8、本发明化合物8的制备
[0087][0088]
制备方法为:在50ml三口烧瓶中加入2.26g(0.010mol)3,4,5-三甲氧基苯乙酸、1.83g(0.012mol)5-氟-3-羟基-4-甲氧基苯甲醛,再加入3.2ml醋酸酐和1.6ml三乙胺,摇匀,装入磁力搅拌子,装上回流冷凝管,通入氮气保护,油浴加热升温到150℃,保温反应5小
时,冷却至80℃,将回流冷凝管换为滴液漏斗,滴入8ml盐酸溶液,保持80℃反应1小时,冷却至室温,滴入40%氢氧化钠溶液2ml,继续反应1小时,然后滴入1.8ml浓盐酸,析出固体之后继续搅拌反应1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体2.45g(收率64.7%)。类白色结晶性固体为(e)-3-(5-氟-3-羟基-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸。
[0089]
在10ml三口烧瓶中加入1.74g(4.84mmol)(e)-3-(5-氟-3-羟基-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸、0.37g(5.08mmol)叔丁胺,10ml无水乙醇,摇匀,装入磁力搅拌子,搅拌,析出固体后,继续搅拌1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体(化合物8)1.52g(收率69.6%)。[m+h]
+
:452.20。1h-nmr(cdcl3,δ(ppm)):1.287(9h),3.635(3h),3.831(9h),6.228(2h),7.267(1h),7.338(1h),7.643(1h)。
[0090]
实施例9、本发明化合物9的制备
[0091][0092]
制备方法为:在50ml三口烧瓶中加入2.26g(0.010mol)3,4,5-三甲氧基苯乙酸、1.83g(0.012mol)2-氟-3-羟基-4-甲氧基苯甲醛,再加入3.2ml醋酸酐和1.6ml三乙胺,摇匀,装入磁力搅拌子,装上回流冷凝管,通入氮气保护,油浴加热升温到150℃,保温反应5小时,冷却至80℃,将回流冷凝管换为滴液漏斗,滴入8ml盐酸溶液,保持80℃反应1小时,冷却至室温,滴入40%氢氧化钠溶液12ml,继续反应1小时,然后滴入10ml浓盐酸,析出固体之后继续搅拌反应1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体2.56g(收率67.6%)。类白色结晶性固体为(e)-3-(2-氟-3-羟基-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸。
[0093]
在10ml三口烧瓶中加入1.74g(4.84mmol)(e)-3-(2-氟-3-羟基-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸、0.37g(5.08mmol)正丁胺,10ml无水乙醇,摇匀,装入磁力搅拌子,搅拌,析出固体后,继续搅拌1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体(化合物9)1.78g(收率81.5%)。[m+h]
+
:452.20。1h-nmr(cdcl3,δ(ppm)):0.912(3h),1.276(2h),2.314(2h),3.251(2h),3.586(3h),3.773(9h),6.643(2h),7.084(1h),7.237(1h),7.539(1h)。
[0094]
实施例10、本发明化合物10的制备
[0095][0096]
制备方法为:在50ml三口烧瓶中加入2.26g(0.010mol)3,4,5-三甲氧基苯乙酸、1.99g(0.012mol)4-氟-3,5-二甲氧基苯甲醛,再加入3.2ml醋酸酐和1.6ml三乙胺,摇匀,装入磁力搅拌子,装上回流冷凝管,通入氮气保护,油浴加热升温到150℃,保温反应5小时,冷却至80℃,将回流冷凝管换为滴液漏斗,滴入8ml盐酸溶液,保持80℃反应1小时,冷却至室温,滴入40%氢氧化钠溶液2ml,继续反应1小时,然后滴入1.8ml浓盐酸,析出固体之后继续搅拌反应1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体2.47g(收率62.9%)。类白色结晶性固体为(e)-3-(3,5-二甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸。
[0097]
在10ml三口烧瓶中加入1.81g(4.84mmol)(e)-3-(3,5-二甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸、10ml无水乙醇,40%氢氧化钾溶液0.7ml,摇匀,装入磁力搅拌子,搅拌,析出固体后,继续搅拌1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体(化合物10)1.45g(收率69.6%)。[m+h]
+
:432.08。1h-nmr(cdcl3,δ(ppm)):3.541(3h),3.682(3h),3.773(9h),6.327(2h),6.495(1h),6.676(1h),7.329(1h)。
[0098]
实施例11、本发明化合物11的制备
[0099][0100]
制备方法为:在50ml三口烧瓶中加入2.26g(0.010mol)3,4,5-三甲氧基苯乙酸、2.02g(0.012mol)3,5-二羟基-4-甲氧基苯甲醛,再加入3.2ml醋酸酐和1.6ml三乙胺,摇匀,装入磁力搅拌子,装上回流冷凝管,通入氮气保护,油浴加热升温到150℃,保温反应5小时,冷却至80℃,将回流冷凝管换为滴液漏斗,滴入8ml盐酸溶液,保持80℃反应1小时,冷却至室温,滴入40%氢氧化钠溶液2ml,继续反应1小时,然后滴入1.8ml浓盐酸,析出固体之后继
续搅拌反应1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体2.14g(收率56.8%)。类白色结晶性固体为(e)-3-(3,5-二羟基-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸。
[0101]
在10ml三口烧瓶中加入1.82g(4.84mmol)(e)-3-(3,5-二羟基-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸、0.37g(5.08mmol)正丁胺,10ml无水乙醇,摇匀,装入磁力搅拌子,搅拌,析出固体后,继续搅拌1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体(化合物11)1.56g(收率71.7%)。[m+h]
+
:450.21。1h-nmr(cdcl3,δ(ppm)):0.821(3h),1.327(2h),2.172(2h),3.319(2h),3.591(3h),3.836(9h),5.243(2h),6.385(2h),7.326(2h),7.613(1h)。
[0102]
实施例12、本发明化合物12的制备
[0103][0104]
制备方法为:在50ml三口烧瓶中加入2.26g(0.010mol)3,4,5-三甲氧基苯乙酸、2.19g(0.012mol)3-羟基-2,4-二甲氧基苯甲醛,再加入3.2ml醋酸酐和1.6ml三乙胺,摇匀,装入磁力搅拌子,装上回流冷凝管,通入氮气保护,油浴加热升温到150℃,保温反应5小时,冷却至80℃,将回流冷凝管换为滴液漏斗,滴入8ml盐酸溶液,保持80℃反应1小时,冷却至室温,滴入40%氢氧化钠溶液2ml,继续反应1小时,然后滴入1.8ml浓盐酸,析出固体之后继续搅拌反应1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体2.38g(收率61.0%)。类白色结晶性固体为(e)-3-(3-羟基-2,4-二甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸。
[0105]
在10ml三口烧瓶中加入1.89g(4.84mmol)(e)-3-(3-羟基-2,4-二甲氧基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸、0.74g(5.08mmol)赖氨酸,再加入15ml乙醇,摇匀,装入磁力搅拌子,水浴加热升温到60℃,溶清后冷却至室温,析出固体后,继续搅拌1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体(化合物12)1.85g(收率75.5%)。[m+h]
+
:537.24。1h-nmr(cdcl3,δ(ppm)):1.183(2h),1.649(2h),2.173(2h),3.286(2h),3.513(1h),3.597(3h),3.876(9h),5.237(2h),5.459(1h),6.525(2h),7.193(1h),7.315(1h),7.613(1h)。
[0106]
实施例13、本发明化合物13的制备
[0107][0108]
制备方法为:在50ml三口烧瓶中加入2.26g(0.010mol)3,4,5-三甲氧基苯乙酸、1.99g(0.012mol)4-乙氧基-3-羟基苯甲醛,再加入3.2ml醋酸酐和1.6ml三乙胺,摇匀,装入磁力搅拌子,装上回流冷凝管,通入氮气保护,油浴加热升温到150℃,保温反应5小时,冷却至80℃,将回流冷凝管换为滴液漏斗,滴入8ml盐酸溶液,保持80℃反应1小时,冷却至室温,滴入40%氢氧化钠溶液2ml,继续反应1小时,然后滴入1.8ml浓盐酸,析出固体之后继续搅拌反应1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体2.02g(收率53.9%)。类白色结晶性固体为(e)-3-(4-乙氧基-3-羟基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸。
[0109]
在10ml三口烧瓶中加入1.81g(4.84mmol)(e)-3-(4-乙氧基-3-羟基苯基)-2-(3,4,5-三甲氧基苯基)丙烯酸、0.74g(5.08mmol)赖氨酸,再加入15ml乙醇,摇匀,装入磁力搅拌子,水浴加热升温到60℃,溶清后冷却至室温,析出固体后,继续搅拌1小时,过滤,滤饼用冰冷的无水乙醇洗涤,减压干燥,获得类白色结晶性固体(化合物13)2.05g(收率81.4%)。[m+h]
+
:521.25。1h-nmr(cdcl3,δ(ppm)):1.187(2h),1.341(3h),1.843(2h),2.117(2h),3.415(2h),3.523(1h),3.786(9h),4.217(2h),5.302(2h),5.476(1h),6.558(2h),7.181(1h),7.328(1h),7.613(1h)。
[0110]
实施例14、本发明化合物制备成0.9%氯化钠注射液
[0111]
制备0.9%氯化钠静脉注射用注射液:
[0112]
处方一:
[0113][0114]
处方二:
[0115][0116]
处方三:
[0117][0118]
在上述处方中,为提高化合物的稳定性,在制备注射液时还可以按照常规方法,分别加入药物中常用的稳定剂、抗氧化剂、ph值调节剂。稳定剂如环糊精包合物、表面活性剂(阴离子表面活性剂、阳离子表面活性剂、两性离子表面活性剂、非离子表面活性剂)等;抗氧化剂如亚硫酸钠、亚硫酸氢钠、焦亚硫酸钠、硫代硫酸钠、抗坏血酸、半胱氨酸等;ph值调节剂如柠檬酸、富马酸、谷氨酸、l-天冬氨酸、乳酸、乳糖酸、半乳糖醛酸、葡萄糖醛酸、抗坏血酸、盐酸、醋酸等。
[0119]
实施例15、本发明化合物制备成无菌粉针剂
[0120]
处方一:
[0121][0122][0123]
处方二:
[0124][0125]
处方三:
[0126][0127]
实施例16、本发明化合物制备成5%葡萄糖注射液
[0128]
制备5%葡萄糖静脉注射用注射液:
[0129]
处方一:
[0130][0131]
处方二:
[0132][0133]
处方三:
[0134][0135]
实施例17、本发明化合物制备成片剂
[0136]
处方:
[0137][0138]
制备片剂时,填充剂可选用如淀粉、糊精、糖粉、预胶化淀粉、乳糖、葡萄糖、微晶纤维素、碳酸钙、硫酸钙、碳酸氢钙等;黏合剂可选用如羟丙甲纤维素、聚维酮、淀粉浆、糊精浆、糖浆、胶浆、海藻酸钠、聚乙二醇、桃胶、阿拉伯胶等;崩解剂可选用如交联羧甲基纤维素钠、交联聚维酮、羧甲基淀粉钠、羟丙基淀粉、低取代羟丙基纤维素柠檬酸、酒石酸、酸酐、碳酸氢钠、碳酸钠等。
[0139]
实施例18、本发明化合物制备成胶囊剂
[0140]
处方:
[0141][0142]
制备胶囊剂时,填充剂可选用如淀粉、糊精、糖粉、预胶化淀粉、乳糖、葡萄糖、微晶纤维素、碳酸钙、硫酸钙、碳酸氢钙等;黏合剂可选用如羟丙甲纤维素、聚维酮、淀粉浆、糊精浆、糖浆、胶浆、海藻酸钠、聚乙二醇、桃胶、阿拉伯胶等。
[0143]
以下通过具体试验例证明本发明的有益效果。
[0144]
试验例1、本发明化合物在小鼠lewis非小细胞肺癌模型中富集杀伤性t淋巴细胞抗原cd8+的研究
[0145]
llc(小鼠肺癌细胞)是小鼠lewis肺癌细胞。
[0146]
llc细胞肺癌小鼠的造模方法为:
[0147]
llc细胞养于含有10%胎牛血清和100μ/ml双抗(青霉素+链霉素)的通用培养基中,在37℃,5%co2的细胞培养箱中常规培养。消化生长至对数期的llc细胞,经0.25%胰蛋白酶(含edta)消化成细胞悬液,1000rpm/min离心3min,弃去上清收集细胞沉淀,用无血清无双抗培养基重悬,通过细胞计数同样条件再次离心调整体积,制成细胞悬浮液,使得细胞浓度为1
×
107/μl。在每只小鼠右腹侧注射200μl细胞悬液,完成肿瘤细胞接种。肿瘤生长约10天以后体积达到100mm3左右开始给药。
[0148]
药物配制:将本发明化合物1~13用注射用水配制成注射液。
[0149]
实验方法:采用腹腔注射的方式对造模成功的llc细胞肺癌小鼠以400mg/kg腹腔注射含本发明化合物的注射液进行治疗,设置不给药的阴性组(造模小鼠仅腹腔注射等体积的注射用水)、给药的实验组(造模小鼠腹腔注射本发明化合物配制的注射液),腹腔注射方法为每天注射1次,在每日固定时间进行给药,给药前称量体重,给药体积10ml/kg,连续给药10天,即接受治疗后第10天,处死小鼠并剥离皮下肿瘤。以流式细胞术检测阴性组、给药组肿瘤组织中杀伤性t淋巴细胞抗原cd8
+
的比例。
[0150]
对肿瘤组织中cd8
+
肿瘤浸润淋巴细胞通过流式细胞仪检测。具体步骤如下:
[0151]
处死小鼠,用眼科剪剪开小鼠腹部表皮,并充分暴露皮下肿瘤,用镊子及眼科剪逐步剥离小鼠右侧腹部的皮下肿瘤组织。用pbs清洗肿瘤组织后,用眼科剪将剥离出的肿瘤组织剪碎至1mm左右,将剪碎的肿瘤组织放入3ml含有100μg/ml透明质酸酶的pbs缓冲溶液中。将装有消化液的离心管放入水浴锅中,37℃水浴20min,每隔5min用移液管吹打震荡消化肿瘤组织,使其充分消化。将包含有未消化完全的肿瘤组织的消化液用70μm不锈钢细胞筛过滤,残留的未被消化的肿瘤组织用20ml针筒的推杆在70μm的细胞筛上研磨,研磨的同时用1ml的pbs冲洗5次。将通过细胞筛的细胞悬液转移至15ml离心管中,4000rpm离心3min后,收集细胞沉淀并加入2ml红细胞裂解液,静置10min后再4000rpm离心3min。使用pbs洗涤后加入2-3ml pbs重悬细胞。
[0152]
取适量细胞作为流式分析样品,对细胞悬液计数并调整细胞浓度至5
×
106/ml,用
移液枪吸取100μl细胞悬液加入流式管。在每一支流式管中分别加入fitc anti mouse cd3、pe-cy5 anti-mouse cd8、pe/cyanine7 anti-mouse cd45,其中前二者同时加入一支流式管中,加入荧光抗体后将流式管重新放置在冰上,避光孵育20min。再流式管中加入2ml灭菌pbs,1000rpm离心5min,弃上清。加入0.5ml灭菌pbs重悬细胞,细胞总量选取为1
×
106,流式细胞仪上机检测,用bd-facsdiva分析结果。
[0153]
比较含有cd8
+
抗原的细胞毒性t淋巴细胞在cd45
+
、cd3
+
和cd8
+
细胞中的比例。
[0154]
通过imagej的ihc profiler插件对肿瘤免疫组化显微图像进行处理,对染色图像进行颜色去卷积,分离出蓝色的阴性区域和棕色的阳性区域,获得阳性区域所占染色区域总面积的百分比。
[0155]
药物对杀伤性淋巴细胞cd8
+
在肿瘤组织中的分布的影响以及杀伤肿瘤细胞作用通过免疫组化进行检测。具体操作步骤如下:
[0156]
在接受治疗10天后处死小鼠,用眼科剪剪开小鼠腹部表皮,切除肿瘤,并放置于福尔马林进行固定。固定完成后用石蜡进行包埋,切割两个连续的3μm切片,安装在载玻片上并干燥(60℃,1h)。分别加入一抗后4℃过夜,滴加二抗。对氨基联苯胺(dab)显色,苏木精复染、脱水、透明后封片。
[0157]
以20倍放大率捕获的整个载玻片图像,在imagej中对切片染色图像进行处理,分出蓝色的阴性区域和棕色的阳性区域,并计算阳性区域占切片总面积的百分比。
[0158]
研究结果如表2所示。
[0159]
表2.本发明化合物对cd8
+
在肿瘤组织中富集的影响及分布面积比例的结果
[0160]
化合物肿瘤组织中cd8
+
在总免疫细胞中所占比例(%)肿瘤组织中分布比例(%)10.0605
±
0.01246.265
±
0.367320.0823
±
0.01127.147
±
0.752830.0762
±
0.01036.731
±
0.569240.0787
±
0.01226.872
±
0.763350.0833
±
0.01077.438
±
0.761060.0814
±
0.01187.249
±
0.668970.0774
±
0.01156.653
±
0.779180.0735
±
0.01276.543
±
0.725690.0753
±
0.01236.258
±
0.6573100.0623
±
0.01145.847
±
0.6349110.0653
±
0.01225.735
±
0.5952120.0753
±
0.01035.883
±
0.6723130.0723
±
0.01266.547
±
0.5843空白0.0417
±
0.01082.125
±
0.2404
[0161]
实验结果说明:本发明化合物具有富集杀伤性t淋巴细胞抗原cd8
+
于肿瘤组织中和靶向破坏肿瘤血管内壁双重作用。可以大大提高杀伤性t淋巴细胞抗原cd8
+
在肿瘤组织中富集比例及分布面积,在通过杀伤性t淋巴细胞抗原cd8
+
消灭肿瘤细胞的同时,切断肿瘤组织的血供,造成实体瘤内部的快速坏死。
[0162]
试验例2、本发明化合物对非融合状态人脐静脉内皮细胞(huvec)的黏附蛋白的影
响
[0163]
融合状态的huvecs:取处于对数生长期的huvecs细胞,按8
×
104个/ml 100μ1/孔接种于96孔培养板(即8000个/孔),细胞生长于含10%胎牛血清的培养液中,置37℃,5%co2培养箱内培养,24h后加入受试化合物。受试样品用pbs(-)稀释后加入各孔内(10μl/孔),同一浓度设3复孔。置培养箱中培养24小时后,每孔分别加入10μl cck-8溶液,注意不要产生气泡。继续放入培养箱内培养2.5小时左右后可测吸光度。用mk-2全自动酶标仪在450nm波长下测定od值,ic50值。
[0164]
非融合状态的huvecs:取处于对数生长期的huvecs细胞,按2
×
104个/ml 100μl/孔接种于96孔培养板(即2000个/孔),细胞生长于含10%胎牛血清的培养液中,置37℃,5%co2培养箱内培养,24h后加入受试化合物。受试样品用pbs(-)稀释后加入各孔内(10μl/孔),同一浓度设3复孔。置培养箱中培养24小时后,每孔分别加入10μl cck-8溶液,注意不要产生气泡。继续放入培养箱内培养2.5小时左右后可测吸光度。用mk-2全自动酶标仪在450nm波长下测定od值,ic50值。
[0165]
以非融合状态人脐静脉内皮细胞(huvec)模拟新生肿瘤血管,而融合状态huvec模拟正常血管,制备实验模型。并以相同剂量10μm化合物处理两种不同状态的人脐静脉内皮细胞,模拟融合细胞状态,制备蛋白质组样品,制备肽段后用tmt标记,开展定量蛋白质组学研究。tmt标记效率高于99.4%,以1%fdr严格卡值,共鉴定到来自4934个蛋白的28372条肽段,其中可定量蛋白4909个,质量可靠。将非融合状态和融合状态药物处理组与各自对照组相比,以对照组为参考,大于1.5倍差异或小于0.67倍差异为显著差异蛋白卡值标准。
[0166]
分别提取非融合细胞组,融合细胞组样品的总细胞蛋白,对蛋白样品取2μg进行质量检测,开展定量蛋白质组学实验,采用灰度定量完成蛋白浓度检测。具体流程图如图1所示。
[0167]
所得数据分析:从uniprot下载人类蛋白数据库(scientific name:homo sapiens,9606)。使用maxquant(v 1.5.5.1)搜索引擎进行搜库。通过tmt定量蛋白质学实验共鉴定到4934个蛋白(4909个定量蛋白),28372条肽段,标记效率高于99%,全酶切比例为86.3%。
[0168]
本发明化合物对非融合状态人脐静脉内皮细胞(huvec)蛋白组学试验结果如表3所示。
[0169]
表3.本发明化合物对蛋白的影响
[0170]
[0171][0172]
将差异蛋白进行go富集分析,非融合组差异表达蛋白主要参与调控细胞间黏附。说明上述化合物靶向破坏非融合状态人脐静脉内皮细胞(huvec)的黏附功能,使内皮细胞从血管基底脱离,从而引起非融合血管炎性反应,进一步造成血管的阻断。
[0173]
试验例3、化合物对人体肺癌a549裸鼠移植瘤的疗效
[0174]
本试验选用移植于裸鼠的人体肺癌a549模型进行受试样品的抗肿瘤作用研究,剂量400mg/kg经口灌胃14天,测量对人体肺癌a549的抑瘤率。
[0175]
配制:用蒸馏水将化合物配至所需浓度,给药前配制。
[0176]
动物品系:balb/c裸鼠(spf级),体重:19-21g,性别:雄性。
[0177]
给药组每组动物数:8只;对照组(control)动物数:>12只。
[0178]
试验方法:
[0179]
取生长良好的肺癌a549瘤块,无菌条件下切割成3mm大小的均匀小块,用套管针每只小鼠右腋皮下接种一块,设以下9组,给药组每组8只,对照组大于12只。
[0180]
接种后14天见瘤块体积平均约100mm3,根据肿瘤大小重新分组,淘汰肿瘤过大和过小的动物,每组肿瘤平均体积基本一致,开始给药:给药组连续经口灌胃14天,每天给药1次,每次给药体积为0.5ml/20g体重;对照组给药相同体积的生理盐水。自接种第14天起每周2次用数显电子卡尺测量肿瘤长径a(mm),及相垂直的肿瘤短径b(mm),肿瘤体积计算公式为:tv=ab2/2,相对肿瘤体积计算公式为:rtv=vt/vo,vo为分笼时(即d1)测量所得肿瘤体积,vt为每一次测量时的肿瘤体积。
[0181]
结果判定根据以下公式:
[0182][0183]
试验结果如表4:
[0184]
表4.化合物对肺癌a549瘤块的抑瘤效果
[0185][0186]
与control组比较:*p<0.05,**p<0.01;其他化合物与化合物6相比:
#
p<0.05,
##
p<0.01。
[0187]
上述试验结果说明:本发明部分化合物对肺癌有显著抑制效果,特别是化合物6抑制肺癌的效果最优,可以用于制备治疗肺癌的药物。
[0188]
试验例4、化合物对人体肝癌qgy7703裸鼠移植瘤的疗效
[0189]
本试验选用移植于裸鼠的人体肝癌qgy7703模型进行受试样品的抗肿瘤作用研究,剂量100mg/kg经静脉注射14天,测量对人体肝癌qgy7703的抑瘤率。
[0190]
配制:用无菌生理盐水将化合物配至所需浓度,给药前配制。
[0191]
动物品系:balb/c裸鼠(spf级);体重:19-21g;性别:雄性。
[0192]
给药组每组动物数:8只;对照组(control)动物数:>12只。
[0193]
试验方法:
[0194]
取生长良好的人体肝癌qgy7703瘤块,无菌条件下切割成3mm大小的均匀小块,用套管针每只小鼠右腋皮下接种一块,设以下9组,给药组每组8只,control组大于12只。
[0195]
接种后14天见瘤块体积平均约100mm3,根据肿瘤大小重新分组,淘汰肿瘤过大和过小的动物,每组肿瘤平均体积基本一致,开始给药:给药连续经静脉注射14天,每天给药1次,每次给药体积为0.1ml/20g体重;对照组给药相同体积的无菌生理盐水。自接种第14天起每周2次用数显电子卡尺测量肿瘤长径a(mm),及相垂直的肿瘤短径b(mm),肿瘤体积计算公式为:tv=ab2/2,相对肿瘤体积计算公式为:rtv=vt/vo,vo为分笼时(即d1)测量所得肿瘤体积,vt为每一次测量时的肿瘤体积。
[0196]
结果判定根据以下公式:
[0197][0198]
试验结果如表5:
[0199]
表5.化合物对肝癌qgy7703瘤块的抑瘤效果
[0200][0201]
与control组比较:*p<0.05,**p<0.01;其他化合物与化合物6相比:
#
p<0.05,
##
p<0.01。
[0202]
上述试验结果说明:本发明部分化合物对肝癌有显著抑制效果,特别是化合物6抑制肝癌的效果最优,可以用于制备治疗肝癌的药物。
[0203]
试验例5、化合物对人体胃癌mgc803裸鼠移植瘤的疗效
[0204]
本试验选用移植于裸鼠的人体胃癌mgc803模型进行受试样品的抗肿瘤作用研究,剂量200mg/kg经腹腔注射14天,测量对人体胃癌mgc803的抑瘤率。
[0205]
配制:用无菌生理盐水将化合物配至所需浓度,给药前配制。
[0206]
动物品系:balb/c裸鼠(spf级);体重:19-21g;性别:雄性。
[0207]
给药组每组动物数:8只;对照组(control)动物数:>12只。
[0208]
试验方法:
[0209]
取生长良好的人体胃癌mgc803瘤块,无菌条件下切割成3mm大小的均匀小块,用套管针每只小鼠右腋皮下接种一块,设以下9组,给药组每组8只,control组大于12只。
[0210]
接种后14天见瘤块体积平均约100mm3,根据肿瘤大小重新分组,淘汰肿瘤过大和过小的动物,每组肿瘤平均体积基本一致,开始给药,给药连续经腹腔注射14天,每天给药1次,每次给药体积为0.2ml/20g体重。自接种第14天起每周2次用数显电子卡尺测量肿瘤长径a(mm),及相垂直的肿瘤短径b(mm),肿瘤体积计算公式为:tv=ab2/2,相对肿瘤体积计算公式为:rtv=vt/vo,vo为分笼时(即d1)测量所得肿瘤体积,vt为每一次测量时的肿瘤体积。
[0211]
结果判定根据以下公式:
[0212][0213]
试验结果如下表6:
[0214]
表6.化合物对胃癌mgc803瘤块的抑瘤效果
[0215][0216]
与control组比较:*p<0.05,**p<0.01;其他化合物与化合物6相比:
#
p<0.05,
##
p<0.01。
[0217]
上述试验结果说明:本发明部分化合物对胃癌有显著抑制效果,特别是化合物6抑制胃癌的效果最优,可以用于制备治疗胃癌的药物。
[0218]
试验例6、化合物对人卵巢癌a2780裸鼠移植瘤的疗效
[0219]
本试验选用移植于裸鼠的人卵巢癌a2780模型进行受试样品的抗肿瘤作用研究,剂量200mg/kg经腹腔注射14天,测量对人卵巢癌a2780的抑瘤率。
[0220]
配制:用无菌生理盐水将化合物配至所需浓度,给药前配制。
[0221]
动物品系:balb/c裸鼠(spf级);体重:19-21g;性别:雄性。
[0222]
给药组每组动物数:8只;对照组(control)动物数:>12只
[0223]
试验方法:
[0224]
取生长良好的人卵巢癌a2780瘤块,无菌条件下切割成3mm大小的均匀小块,用套管针每只小鼠右腋皮下接种一块,设以下9组,给药组每组8只,control组大于12只。
[0225]
接种后14天见瘤块体积平均约100mm3,根据肿瘤大小重新分组,淘汰肿瘤过大和过小的动物,每组肿瘤平均体积基本一致,开始给药:给药连续经腹腔注射14天,每天给药1次,每次给药体积为0.2ml/20g体重;对照组给药相同体积的无菌生理盐水。自接种第14天起每周2次用数显电子卡尺测量肿瘤长径a(mm),及相垂直的肿瘤短径b(mm),肿瘤体积计算公式为:tv=ab2/2,相对肿瘤体积计算公式为:rtv=vt/vo,vo为分笼时(即d1)测量所得肿瘤体积,vt为每一次测量时的肿瘤体积。
[0226]
结果判定根据以下公式:
[0227][0228]
试验结果如下表7:
[0229]
表7.化合物对卵巢癌a2780瘤块的抑瘤效果
[0230][0231]
与control组比较:*p<0.05,**p<0.01;其他化合物与化合物6相比:#p<0.05,##p<0.01。
[0232]
上述试验结果说明:本发明部分化合物对卵巢癌有显著抑制效果,特别是化合物6抑制卵巢癌的效果最优,可以用于制备治疗卵巢癌的药物。
[0233]
试验例7、化合物对人前列腺细胞lncap裸鼠移植瘤的疗效
[0234]
本试验选用移植于裸鼠的人前列腺细胞lncap模型进行受试样品的抗肿瘤作用研究,剂量200mg/kg经腹腔注射14天,测量对人前列腺细胞lncap的抑瘤率。
[0235]
配制:用无菌生理盐水将化合物配至所需浓度,给药前配制。
[0236]
动物品系:balb/c裸鼠(spf级);体重:19-21g;性别:雄性。
[0237]
给药组每组动物数:8只;对照组(control)动物数:>12只
[0238]
试验方法:
[0239]
取生长良好的人前列腺细胞lncap瘤块,无菌条件下切割成3mm大小的均匀小块,用套管针每只小鼠右腋皮下接种一块,设以下9组,给药组每组8只,control组大于12只。
[0240]
接种后14天见瘤块体积平均约100mm3,根据肿瘤大小重新分组,淘汰肿瘤过大和过小的动物,每组肿瘤平均体积基本一致,开始给药:给药连续经腹腔注射14天,每天给药1次,每次给药体积为0.2ml/20g体重;对照组给药相同体积的无菌生理盐水。自接种第14天起每周2次用数显电子卡尺测量肿瘤长径a(mm),及相垂直的肿瘤短径b(mm),肿瘤体积计算公式为:tv=ab2/2,相对肿瘤体积计算公式为:rtv=vt/vo,vo为分笼时(即d1)测量所得肿瘤体积,vt为每一次测量时的肿瘤体积。
[0241]
结果判定根据以下公式:
[0242][0243]
试验结果如下表8:
[0244]
表8.化合物对前列腺细胞lncap瘤块的抑瘤效果
[0245][0246][0247]
与control组比较:*p<0.05,**p<0.01;其他化合物与化合物4相比:
#
p<0.05,
##
p<0.01。
[0248]
上述试验结果说明:本发明部分化合物对前列腺癌有显著抑制效果,特别是化合物4抑制前列腺癌的效果最优,可以用于制备治疗前列腺癌的药物。
[0249]
试验例8、化合物对人肾细胞癌aghn裸鼠移植瘤的疗效
[0250]
本试验选用移植于裸鼠的人肾细胞癌aghn模型进行受试样品的抗肿瘤作用研究,剂量200mg/kg经腹腔注射14天,测量对人肾细胞癌aghn的抑瘤率。
[0251]
配制:用无菌生理盐水将化合物配至所需浓度,给药前配制。
[0252]
动物品系:balb/c裸鼠(spf级);体重:19-21g;性别:雄性。
[0253]
给药组每组动物数:8只;对照组动物数:>12只。
[0254]
试验方法:
[0255]
取生长良好的人肾细胞癌aghn瘤块,无菌条件下切割成3mm大小的均匀小块,用套管针每只小鼠右腋皮下接种一块,设以下9组,给药组每组8只,control组大于12只。
[0256]
接种后14天见瘤块体积平均约100mm3,根据肿瘤大小重新分组,淘汰肿瘤过大和过小的动物,每组肿瘤平均体积基本一致,开始给药:给药连续经腹腔注射14天,每天给药1次,每次给药体积为0.2ml/20g体重;对照组给药相同体积的无菌生理盐水。自接种第14天起每周2次用数显电子卡尺测量肿瘤长径a(mm),及相垂直的肿瘤短径b(mm),肿瘤体积计算公式为:tv=ab2/2,相对肿瘤体积计算公式为:rtv=vt/vo,vo为分笼时(即d1)测量所得肿瘤体积,vt为每一次测量时的肿瘤体积。
[0257]
结果判定根据以下公式:
[0258][0259]
试验结果如下表9:
[0260]
表9.化合物对肾细胞癌aghn瘤块的抑瘤效果
[0261][0262][0263]
与control组比较:*p<0.05,**p<0.01;其他化合物与化合物4相比:
#
p<0.05,
##
p<0.01。
[0264]
上述试验结果说明:本发明部分化合物对肾细胞癌有显著抑制效果,特别是化合物4抑制肾细胞癌的效果最优,可以用于制备治疗肾细胞癌的药物。
[0265]
试验例9、化合物6引起体内肿瘤坏死
[0266]
1、试验方法:
[0267]
取生长良好的实体瘤(人体肺癌95d和人体肝癌bel7404),无菌条件下切割成约2-3mm大小的均匀小块,用套管针每只裸鼠右腋皮下接种一块。接种后14天口服给药(化合物6),给药剂量为400mg/kg,给药体积为0.5ml/20g体重,药物使用无菌生理盐水配制。给药后不同时间点(4h、24h),处死裸鼠解剖取肿瘤组织,用10%福尔马林固定,做病理切片,he染色,拍照,观察肿瘤内部坏死情况,细胞增殖情况。
[0268]
2、试验结果:
[0269]
he染色如图2和图3所示:化合物6口服400mg/kg给药后4小时肿瘤组织内部开始出现坏死,24小时可以导致肿瘤内部大面积的坏死(浅色区域)。说明本发明化合物6能够有效引起肿瘤坏死,达到抗肿瘤的目的。
[0270]
试验例10、化合物6引起体内肿瘤内部血管坏死
[0271]
1、试验方法:
[0272]
取生长良好的实体瘤(人体肺癌95d和人体肝癌bel7404),无菌条件下切割成约2-3mm大小的均匀小块,用套管针每只裸鼠右腋皮下接种一块。接种后14天口服给药(化合物6),给药剂量为400mg/kg,给药体积为0.5ml/20g体重,药物使用无菌生理盐水配制。给药后不同时间点(4h、24h),处死裸鼠解剖取肿瘤组织,用10%福尔马林固定,做病理切片,pcna免疫组化染色,拍照,观察肿瘤内瘤血管破坏情况。
[0273]
2、试验结果:
[0274]
pcna免疫组化显示(图4和图5):化合物6口服400mg/kg给药后4h肿瘤组织内的仅存在部分血管内皮细胞的增殖,给药后24h可以引起肿瘤内部大范围血流的阻滞和肿瘤组织坏死。肿瘤组织中pcna表达下调,表明其体内较好的肿瘤血管破坏作用。
[0275]
试验例11、化合物3联合伦伐替尼对人肝癌bel7404裸鼠异种移植肿瘤生长抑制作
用
[0276]
试验方法:人肝癌bel7404裸小鼠移植瘤,由人肝癌bel7404细胞株接种于裸小鼠腋窝皮下而建立。细胞接种量为1
×
106,接种形成移植瘤后再在裸小鼠体内传3代后使用。取生长旺盛期的瘤组织剪切成1.5mm3左右,在无菌条件下匀浆后制备成1
×
107/ml细胞悬液,以0.1ml接种于裸小鼠右侧腋窝皮下。裸小鼠移植瘤用游标卡尺测量移植瘤直径,待肿瘤生长至100mm3后将动物随机分组,每组6只。使用测量瘤径的方法,动态观察被试物抗肿瘤的效应。化合物3以400mg/kg,口服给药,每天给药一次,给药三周;伦伐替尼10mg/kg口服给药,每天给药一次,给药两周。肿瘤直径的测量次数为每三天测一次。阴性对照组口服等量生理盐水溶液。合并用药的效应根据金氏公式计算q值:
[0277]
q=ea+b/(ea+eb-ea
×
eb)
[0278]
ea+b为合并用药的抑瘤率,ea和eb分别为a药和b药的抑瘤率。如q值0.85~1.15为相加(+),>1.15为增强(++)。
[0279]
试验结果如下表10:
[0280]
表10.化合物3联合伦伐替尼对人肝癌bel7404裸鼠异种移植肿瘤生长抑制作用
[0281][0282]
与空白对照组比较,
*
p<0.05
[0283]
试验结论:
[0284]
化合物3合并伦伐替尼的q值为0.970,有明显的抗肿瘤相加作用,但没有协同增效的抗肿瘤作用。动物体重无明显降低,合并伦伐替尼,也未见明显毒性相加反应。
[0285]
试验例12、化合物6联合紫杉醇对人体胃癌sgc7901裸鼠异种移植肿瘤生长抑制作用
[0286]
试验方法:人体胃癌sgc7901裸小鼠移植瘤,由人体胃癌sgc7901细胞株接种于裸小鼠腋窝皮下而建立。细胞接种量为1
×
106,接种形成移植瘤后再在裸小鼠体内传3代后使用。取生长旺盛期的瘤组织剪切成1.5mm3左右,在无菌条件下匀浆后制备成1
×
107/ml细胞悬液,以0.1ml接种于裸小鼠右侧腋窝皮下。裸小鼠移植瘤用游标卡尺测量移植瘤直径,待肿瘤生长至100mm3后将动物随机分组,每组6只。使用测量瘤径的方法,动态观察被试物抗肿瘤的效应。化合物6以200mg/kg,腹腔注射给药,每隔一天给药一次,给药三周;紫杉醇以10mg/kg静脉注射给药,每隔一天给药一次,给药三周。肿瘤直径的测量次数为每三天测一次。给药体积为0.1ml/20g。阴性对照组静脉注射等量生理盐水溶液。合并用药的效应根据金氏公式计算q值:
[0287]
q=ea+b/(ea+eb-ea
×
eb)
[0288]
ea+b为合并用药的抑瘤率,ea和eb分别为a药和b药的抑瘤率。如q值0.85~1.15为相加(+),>1.15为增强(++)。
[0289]
试验结果如下表11:
[0290]
表11.化合物6联合紫杉醇对人体胃癌sgc7901裸鼠异种移植肿瘤生长抑制作用
[0291][0292]
与空白对照组比较,
*
p<0.05
[0293]
试验结论:
[0294]
化合物6合并紫杉醇的q值为0.927,有明显的抗肿瘤相加作用,但没有协同增效的抗肿瘤作用。动物体重无明显降低,合并紫杉醇,也未见明显毒性相加反应。
[0295]
试验例13、化合物8联合卡铂对人体肺癌a549裸鼠异种移植肿瘤生长抑制作用
[0296]
试验方法:人体肺癌a549裸小鼠移植瘤,由人体肺癌a549细胞株接种于裸小鼠腋窝皮下而建立。细胞接种量为1
×
106,接种形成移植瘤后再在裸小鼠体内传3代后使用。取生长旺盛期的瘤组织剪切成1.5mm3左右,在无菌条件下匀浆后制备成1
×
107/ml细胞悬液,以0.1ml接种于裸小鼠右侧腋窝皮下。裸小鼠移植瘤用游标卡尺测量移植瘤直径,待肿瘤生长至100mm3后将动物随机分组,每组6只。使用测量瘤径的方法,动态观察被试物抗肿瘤的效应。化合物8以200mg/kg,腹腔注射给药,每隔一天给药一次,给药三周;卡铂以10mg/kg腹腔注射给药,每隔一天给药一次,给药三周。肿瘤直径的测量次数为每三天测一次。给药体积为0.1ml/20g。阴性对照组静脉注射等量生理盐水溶液。合并用药的效应根据金氏公式计算q值:
[0297]
q=ea+b/(ea+eb-ea
×
eb)
[0298]
ea+b为合并用药的抑瘤率,ea和eb分别为a药和b药的抑瘤率。如q值0.85~1.15为相加(+),>1.15为增强(++)。
[0299]
试验结果如下表12:
[0300]
表12.化合物8联合卡铂对人体肺癌a549裸鼠异种移植肿瘤生长抑制作用
[0301][0302]
与空白对照组比较,
*
p<0.05
[0303]
试验结论:
[0304]
化合物8合并卡铂的q值为0.941,有明显的抗肿瘤相加作用,但没有协同增效的抗肿瘤作用。动物体重无明显降低,合并卡铂,也未见明显毒性相加反应。
[0305]
试验例14、化合物11联合pd-l1对小鼠lewis非小细胞肺癌模型中的肿瘤抑制作用
[0306]
试验方法:c57bl/6小鼠移植瘤,由lewis非小细胞肺癌株接种于c57bl/6小鼠腋窝皮下而建立。细胞接种量为1
×
106,接种形成移植瘤后再在c57bl/6小鼠体内传3代后使用。取生长旺盛期的瘤组织剪切成1.5mm3左右,在无菌条件下匀浆后制备成1
×
107/ml细胞悬液,以0.1ml接种于c57bl/6小鼠右侧腋窝皮下。c57bl/6小鼠移植瘤用游标卡尺测量移植瘤直径,待肿瘤生长至100mm3后将动物随机分组,每组6只。使用测量瘤径的方法,动态观察被试物抗肿瘤的效应。化合物11以200mg/kg,腹腔注射给药,每隔一天给药一次,给药三周;pd-l1以10mg/kg腹腔注射给药,每隔一天给药一次,给药三周。肿瘤直径的测量次数为每三天测一次。给药体积为0.1ml/20g。阴性对照组静脉注射等量生理盐水溶液。
[0307]
合并用药的效应根据金氏公式计算q值:
[0308]
q=ea+b/(ea+eb-ea
×
eb)
[0309]
ea+b为合并用药的抑瘤率,ea和eb分别为a药和b药的抑瘤率。如q值0.85~1.15为相加(+),>1.15为增强(++)。
[0310]
试验结果如下表13:
[0311]
表13.化合物11联合pd-l1对小鼠lewis非小细胞肺癌模型中的肿瘤抑制作用
[0312][0313]
与空白对照组比较,
*
p<0.05
[0314]
试验结论:
[0315]
化合物11合并pd-l1的q值为1.16,说明化合物11与pd-l1联合使用有明显的协同增效抗肺癌作用。动物体重无明显降低,合并用药,也未见明显毒性相加反应。
[0316]
试验例15、化合物6联合t细胞对nog小鼠人体肝癌qgy7703模型中的肿瘤抑制作用
[0317]
试验方法:qgy7703肿瘤细胞用pbs洗涤两次,然后用pbs:matrigel(体积比1∶1混合)重悬,调整细胞浓度为2.5
×
106个/ml,接种于实验动物的右侧腋下皮下,200μl/小鼠,即5
×
105个细胞/小鼠。肿瘤生长至50mm3左右时分组给药。
[0318]
t细胞培养及收获:供者的pbmc,根据t细胞培养标准操作规程进行体外活化扩增。收获对数生长期细胞,按照中剂量(2
×
107个/ml)要求悬浮于溶媒中备用。
[0319]
当肿瘤生长至适宜大小时,进行t细胞注射和化合物6用药,该时间记为d0。化合物6给药方式为腹腔注射,剂量为200mg/kg。d0天开始给药,每天给药,持续给药14天。t细胞给药方式为皮下给药,且给药部位与肿瘤细胞接种部位相同,d0给药,分三次注射,注射方案见下表14。t细胞每次给药在化合物6给药后4小时左右进行。
[0320]
每3天记录小鼠的一般症状、小鼠体重、肿瘤体积和肿瘤重量。基于肿瘤的状态,决定是否追加eal注射。
[0321]
表14.给药方案
[0322]
组别注射途径eal浓度(个/ml)注射体积(ml)eal用量(个/只)
d0给药皮下注射2
×
1070.12
×
106d1给药皮下注射2
×
1070.24
×
106d2给药皮下注射2
×
1070.24
×
106[0323]
合并用药的效应根据金氏公式计算q值:
[0324]
q=ea+b/(ea+eb-ea
×
eb)
[0325]
ea+b为合并用药的抑瘤率,ea和eb分别为a药和b药的抑瘤率。如q值0.85~1.15为相加(+),>1.15为增强(++)。
[0326]
试验结果如下表15:
[0327]
表15.化合物6联合t细胞对nog小鼠人体肝癌qgy7703模型中的肿瘤抑制作用
[0328][0329]
与空白对照组比较,
*
p<0.05
[0330]
试验结论:
[0331]
化合物6合并t细胞的q值为1.20,说明化合物6与t细胞联合使用有明显的协同增效抗肝癌作用。动物体重无明显降低,合t细胞,也未见明显毒性相加反应。
[0332]
试验例14和15说明本发明二苯乙烯类化合物可以与肿瘤免疫治疗药物联合使用,发挥协同增效抗肿瘤功效。
[0333]
综上,本发明提供了二苯乙烯类化合物在制备抗肿瘤药物中的用途,本发明研究发现部分二苯乙烯类化合物具有富集实体瘤内t淋巴细胞cd8
+
及靶向破坏肿瘤血管内壁的双重作用,在通过杀伤性t淋巴细胞抗原cd8
+
消灭肿瘤细胞的同时,切断肿瘤组织的血供,造成实体瘤内部的快速坏死,大大提高对肿瘤的杀伤力。本发明二苯乙烯类化合物可用于制备抗肿瘤药物,也可以与肿瘤免疫治疗药物联合使用,发挥协同增效抗肿瘤功效。这些二苯乙烯类化合物中不同结构的化合物抗肿瘤功效不同,可用于制备不同的抗肿瘤药物,具有良好的应用前景。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1