一种四氧化三锰纳米颗粒的制备及应用
1.本发明涉及一种四氧化三锰纳米颗粒的制备方法,本发明同时还涉及该四氧化三锰纳米颗粒纳在制备聚多巴胺纳米点,以及在检测多巴胺中的应用。
背景技术:
2.多巴胺作为一种重要的儿茶酚胺类神经递质,在心血管、内分泌系统和中枢神经系统的功能中发挥着至关重要的作用。一些重要的神经系统疾病,如:精神分裂症、阿尔茨海默病、帕金森病等均与多巴胺的功能失调有关。因此,多巴胺浓度变化的准确测定对研究神经系统中多巴胺相关的生理病理过程具有十分重要的意义。现有的高灵敏多巴胺检测方法一般是将样品分离技术与分析检测技术相结合,但这些方法由于仪器和操作条件比较复杂、时空分辨率低,使其受到了一定的限制。同时,在多巴胺的检测过程中,由于抗坏血酸、尿酸等于多巴胺具有极其相似的结构和性质,所以在实际应用过程经常会产生基质干扰。因此,制备具有理想分析性能的新型纳米材料来构建荧光传感器用于多巴胺的高选择性检测就显得尤为重要。
技术实现要素:
3.本发明的目的是提供一种四氧化三锰纳米颗粒的制备方法;本发明的另一目的是提供四氧化三锰纳米颗粒在类芬顿反应中用于聚多巴胺纳米点的制备;本发明还有一个目的,就是提供四氧化三锰纳米颗粒在检测多巴胺中的应用。
4.(一)四氧化三锰纳米颗粒的制备本发明制备四氧化三锰纳米颗粒的方法:将硝酸锰加入到碱性低共熔溶剂中,在搅拌下逐滴加入超纯水,室温下静置反应 15~60 min,得棕色沉淀粗产品;粗产品反复用水和无水乙醇洗涤至中性,干燥,即得四氧化三锰纳米颗粒。
5.所述碱性低共熔溶剂是将naoh或koh加热溶解于peg
‑
200中得到的澄清透明溶液,且naoh或koh与peg
‑
200的摩尔比为1:40~1:50,优选1:44。硝酸锰以0.05~0.5 g/ml加入到碱性低共熔溶剂中。
6.加入超纯水的体积为碱性低共熔溶剂体积的0.5~1.5倍。
7.图1为上述制备的四氧化三锰纳米颗粒的xrd图,从图中可以看出,材料的系列衍射峰分别对应于四氧化三锰的(101),(112),(200),(103),(211),(004),(220),(204),(105),(312),(303),(321),(224)和 (400)晶面,这与卡片四氧化三锰的jcpds no:24
‑
734完全吻合,证实了制得的材料为四氧化三锰。
8.图2a为四氧化三锰纳米颗粒的透射电镜(tem)图,从图中可以看出,四氧化三锰纳米颗粒的平均粒径大约为20 nm。图2b为四氧化三锰纳米颗粒的hrtem图,从图中可以看出,材料具有明显的晶格条纹,并且条纹间距为4.9 a
o
,这与四氧化三锰的(101)晶面完全吻合。
9.图3为四氧化三锰纳米颗粒的拉曼光谱图。在351 cm
−1和644 cm
−1处的谱带证实了材料确实为四氧化三锰纳米颗粒。
10.(二)聚多巴胺纳米点的制备将上述制备的四氧化三锰纳米颗粒分散于缓冲溶液中,加入双氧水、多巴胺,于室温下反应0.5~2 h,加入ph=4. 0的醋酸缓冲溶液终止反应;然后通过外加磁场分离并收集上清液,将上清液装入透析袋透析后,冷冻干燥,即得聚多巴胺纳米点粉末。
11.所述缓冲溶液为ph=8.0的4
‑
羟乙基哌嗪乙磺酸溶液或ph=8.0的三羟甲基氨基甲烷缓冲溶液。缓冲溶液中,四氧化三锰纳米颗粒的浓度为10~100
ꢀµ
g/ml,双氧水的摩尔浓度为0.1~200 mm,多巴胺的摩尔浓度为5 mm~100
ꢀµ
m。
12.所述透析袋的截留分子量为500~1000 da,透析时间为12~24 h。
13.图4为上述制备的聚多巴胺纳米点的透射电镜图及粒径分布直方图,其中右上角的小插图为聚多巴胺纳米点的粒径分布直方图,可以看到聚多巴胺纳米点的平均粒径大约为1.7 nm。
14.图5a为聚多巴胺纳米点的xps全谱,结果表明材料主要由c、n和o三种元素组成。图5b为聚多巴胺纳米点的c1s精细谱,可以看出该材料存在c
‑
c/c=c、c
‑
n/c
‑
o和c=n/c=o 键;图5c为聚多巴胺纳米点的o1s精细谱,可以看出该材料存在c
‑
o和c=o键;图5d为聚多巴胺纳米点的n1s精细谱,可以看出该材料存在n
‑
c和n
‑
h键。该四氧化三锰纳米颗粒能够呈现类芬顿反应,可将双氧水分解为羟基自由基,同时使多巴胺在羟基自由基的作用下最终生成具有蓝色荧光的聚多巴胺纳米点,其最佳发射波长为488 nm。
15.(三)多巴胺的检测分别取一系列相同质量的四氧化三锰纳米颗粒(0.03 mg)分散于20 mm(300
ꢀµ
l) ph=8.0的4
‑
羟乙基哌嗪乙磺酸缓冲溶液(四氧化三锰纳米颗粒的浓度为100
ꢀµ
g/ml)中,再分别加入一定量的双氧水(300
ꢀµ
l、100 mm)和300
ꢀµ
l不同浓度的多巴胺(浓度依次为0.05,0.1,1,10,50,100,150,180,200,300,400,500,600 和 1000 μm),于室温下反应1 h;随后加入100
ꢀµ
l醋酸缓冲溶液(10 mm、ph=4.0)终止反应。测定体系在488 nm处的荧光强度,即可对多巴胺进行定量检测。
16.图6a为加入不同浓度的多巴胺后体系的荧光发射光谱图。从图6a和图6b中可以看出,随着多巴胺浓度的增加(从下至上多巴胺的浓度逐渐从0.05
ꢀµ
m增加到1000
ꢀµ
m),体系在488 nm处的荧光强度逐渐增强,并在0.05
ꢀµ
m至300
ꢀµ
m之间,多巴胺浓度与体系在488 nm处的荧光强度呈现良好的线性关系(图6b中的插图),线性回归方程为:y=63.9159+1.1904x,其中y为体系在488 nm处的荧光强度,x为多巴胺的浓度。
17.以空白溶液10次测定结果的标准偏差的3倍为信噪比,得出该方法对多巴胺的检测限为0.017
ꢀµ
m,表明该方法具有较宽的线性范围和较低的检测限。
18.图7为体系在不同干扰物存在下的荧光强度柱状图。从图7中可以看出,只有在多巴胺存在的情况下,体系的荧光才会有明显的增强。表明本发明检测多巴胺具有良好的选择性。
19.综上所述,本发明在碱性低共熔溶剂中制备的四氧化三锰纳米颗粒可发生类芬顿反应,用于聚多巴胺纳米点的制备,也可以用于多巴胺的高选择性检测。
附图说明
20.图1为四氧化三锰纳米颗粒的xrd图。
21.图2为四氧化三锰纳米颗粒的透射电镜(tem)图(a)以及高分辨透射电镜(hrtem)图(b)。
22.图3为四氧化三锰纳米颗粒的拉曼光谱图。
23.图4为聚多巴胺纳米点的透射电镜(tem)图及粒径分布直方图。
24.图5为聚多巴胺纳米点的xps全谱及c1s、o1s和n1s的精细谱。
25.图6为加入不同浓度多巴胺后体系的荧光光谱图(a)以及多巴胺的标准曲线图(b)。
26.图7为体系在不同干扰物存在下的荧光强度柱状图。
具体实施方式
27.下面通过具体实施例对本发明中四氧化三锰纳米颗粒的制备方法及其在类芬顿反应中用于聚多巴胺纳米点的制备以及多巴胺的检测做进一步说明。
28.实施例1、四氧化三锰纳米颗粒的制备将500 mg硝酸锰加入到15 ml 的碱性低共熔溶剂(koh与peg
‑
200按照摩尔比1:44组成)中,再在搅拌下逐滴加入10 ml超纯水,在室温下静止反应 0.5 h,得棕色沉淀为粗产品;粗产品反复用水和无水乙醇洗涤至中性后置于70 ℃烘箱中干燥 12 h,得486 mg四氧化三锰纳米颗粒。
29.实施例2、聚多巴胺纳米点的制备取实施例1制备的四氧化三锰纳米颗粒0.05 mg,分散于(20 mm,1 ml) ph=8.0的4
‑
羟乙基哌嗪乙磺酸缓冲溶液(四氧化三锰纳米颗粒的浓度为50
ꢀµ
g/ml)中,加入50 mm双氧水、80 mm多巴胺,于室温下反应1 h;随后加入10 mmph=4.0醋酸缓冲溶液终止反应。反应结束后,通过外加磁场进行分离并收集上清液,并将上清液装入截留分子量为500 da的透析袋透析12 h后,冷冻干燥,即得聚多巴胺纳米点粉末0.043 mg。
30.实施例3、多巴胺标准样品的检测分别取一系列相同质量的四氧化三锰纳米颗粒(0.03 mg)分散于20 mm(300
ꢀµ
l) ph=8.0的4
‑
羟乙基哌嗪乙磺酸缓冲溶液(四氧化三锰纳米颗粒的浓度为100
ꢀµ
g/ml)中,再分别加入一定量的双氧水(300
ꢀµ
l、100 mm)和300
ꢀµ
l不同浓度的多巴胺(浓度依次为0.05,0.1,1,10,50,100,150,180,200,300,400,500,600 和 1000 μm),于室温下反应1 h;随后加入100
ꢀµ
l醋酸缓冲溶液(10 mm、ph=4.0)终止反应。测定体系在488 nm处的荧光强度,构建线性关系:y=63.9159+1.1904x,其中y为体系在488 nm处的荧光强度,x为多巴胺的浓度。
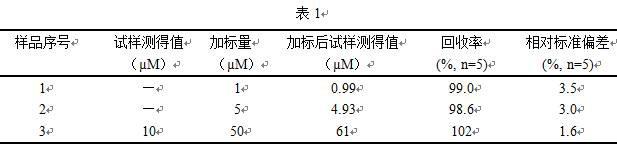
31.实施例4、复杂生物样品中多巴胺的检测取相同体积的盐酸多巴胺注射液作为样品按照多巴胺标准样品的检测过程进行分析,计算测定结果、加标回收率及相对标准偏差,结果如表1所示。其平均回收率在98.6%~102之间,相对标准偏差<5.0%。表明该方法可用于实际样品的检测,且四氧化三锰纳米颗粒用于检测多巴胺具有较高的准确性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1