本发明涉及生物医学,具体是涉及建立导致人类CD36缺失的CD36突变基因真核稳定表达细胞株的方法。
背景技术:
CD36 (白细胞分化抗原36),又名血小板糖蛋白IV(GPIV)、GP88、GPIIIb、 FAT或SCARB3,属于B类清道夫受体跨膜糖蛋白家族。该蛋白分子能被广泛糖基化,分子质量约为88000。人类CD36蛋白由472个氨基酸组成。CD36蛋白在人体内广泛分布于多种细胞和组织中,包括血小板、单核/巨噬细胞、微血管内皮细胞、大脑小胶质细胞、星形胶质细胞、心肌和骨骼肌、脂肪细胞、树突状细胞、视网膜上皮细胞和乳腺及肾脏组织。其主要的功能包括作为凝血酶受体,胶原蛋白受体,长链脂肪酸受体,选择性胆固醇酯摄取受体和溶酶体膜内在蛋白II等受体;还参与诱导细胞凋亡,去除血浆中的氧化低密度脂蛋白,增强异常形态红细胞的黏附等功能。CD36通过与不同配体的结合和与许多配体(I型和IV型胶原,血小板反应蛋白,恶性疟原虫寄生的红细胞,血小板凝集蛋白p37,氧化的低密度脂蛋白和长链脂肪酸等)的相互作用,以及作为细胞表面的黏附分子,在多种生理病理过程中发挥了重要的作用,大量的研究已证明:CD36涉及到止血、血栓、 血小板同种免疫性疾病、高胆固醇血症、肥胖症、外周动脉粥样硬化、动脉高血压、心肌病、糖尿病、疟疾、早老年型痴呆(阿尔茨海默病)等疾病的形成。
部分健康个体在血小板或和单核细胞可出现CD36缺失,按表现型CD36缺失可分为2类:I型CD36缺失,血小板和单核细胞上均不表达CD36;II型CD36缺失,CD36仅在血小板不表达,但在单核细胞表达。CD36缺失的发生率在不同民族、不同地区人群之间的差异很大,在白种人中较罕见(发生率仅为0~0.33%),而在亚洲人群为3%~11%,在撒哈拉以南的非洲人群为8%。本申请人调查(见中国输血杂志,2014年第27卷第1期)发现广西人群(4621名调查个体)CD36缺失的总体发生率是4.13%,其中汉族人群缺失率为3.59%,瑶族为2.38%,而壮族最高为5.76%,CD36 I型缺失的总体比例为41.03%;文献报道的CD36缺失率在深圳地区人群(408名调查个体)为3.19%、广州地区人群(249名调查个体)为2.81%,上海人群(1022名调查个体)为2.15%。
人类 CD36基因的变异,是导致CD36抗原缺失的重要原因。CD36基因位于第7号染色体的q11.2,有15个外显子,长度超过32000bp。目前国际上已报道的可导致CD36蛋白缺失的CD36突变基因的分子基础可大致分为两类:(1)插入或缺失突变:基因突变的结果主要是引起移码突变和外显子跳读。(2)单核苷酸多态性(SNP)突变:结果主要是引起氨基酸置换。目前国际上已报道了近30种可导致CD36蛋白缺失的CD36突变基因,其中本申请人在广西人群中检测到了8个新突变基因,分别是SNP T200A(国际基因序列数据库GenBank号:HM217021.1),SNP C1156T和 SNP T1409C(GenBank号:HM217024.1),SNP T538C(GenBank号:HM217022.1),SNP T1225G(GenBank号:HM217020.1),编码外显子8缺失(导致一段64个氨基酸的蛋白剪切)(GenBank号:HM217025.1),编码外显子 6和7缺失(导致一段40个氨基酸的蛋白剪切)(GenBank号:HM217026.1),编码外显子4缺失(导致一段60个氨基酸的蛋白剪切)(GenBank号:HM217023.1),以及SNP C220T(GenBank号:KF539919)。
大量的研究表明,CD36在人体细胞和组织上表达的量低或缺失,对CD36所涉及的各种疾病的形成和严重程度有很大关系,特别是对疟疾、高胆固醇血症、肥胖症、外周动脉粥样硬化、心肌病、糖尿病、早老年型痴呆等疾病的有直接的影响,其中对CD36缺失个体,CD36蛋白更可作为同种抗原(isoantigen),通过输血、妊娠、骨髓移植等途径,使CD36缺失个体被免疫而产生抗-CD36同种抗体(isoantibody),从而导致包括免疫性的血小板输注无效、输血后紫癜,胎儿/新生儿同种免疫血小板减少症和被动血小板免疫减少症等各类免疫性血小板异常临床问题的发生。本申请人在对到2014年为止公开报道的发生在亚洲人群的635例免疫性的血小板输注无效、输血后紫癜和胎儿/新生儿同种免疫血小板减少症患者的数据进行调查和分析发现(见ISBT Science Series,2014年第9卷第1期):介导这635例血小板免疫异常案例的抗血小板抗体中,除抗-HLA抗体外,抗-CD36抗体检出率为第1位,占2.83%,其中50%的案例是发生在中国人群患者中,证明了抗-CD36在亚洲人群特别是中国人群中,是介导血小板免疫减少症的很重要抗体,也是有别于白种人的特征性血小板同种免疫抗体。
对CD36的表达、调控和参与体内不同生理和病理过程机制正在开展深入的研究,随着对人类疾病认识的提高,CD36无疑将在相关疾病的预防和治疗,以及相关药物的研发中发挥重要作用。在这些研究和应用工作中,导致人类CD36缺失的CD36突变基因和所表达的蛋白,是关键性的十分稀少和珍贵的研究资源及工作平台。
日本Hayashi T等(Hayashi T, Yasui K, Matsuyama N,et al. Establishment of a novel method for detecting Naka antibodies by using a panel cell line.Transfusion, 2009,49(2):390-2. doi: 10.1111/j.1537-2995.),曾应用转染技术(病毒感染法)成功建立了CD36高表达的CD36-K562真核稳定表达细胞株,用于抗-CD36抗体检测的谱细胞系。付丽辉等(付丽辉, 张杰, 孙春昀, 等. 重组人血小板CD36基因真核表达载体的构建及蛋白表达.中国输血杂志, 2012:25(6):557-560.),从人肝组织中提取RNA,反转录CD36 cDNA后成功构建pTE2-s-CD36-10his真核瞬时表达载体,并转染(人工脂质体法)入HEK-293细胞,使成功转染的CD36-HEK-293细胞高表达CD36蛋白,但该法是瞬时转染法,只能使成功转染的CD36-HEK-293细胞株瞬时表达CD36,而不能永久稳定表达CD36。以上两种方法均是正常CD36基因的真核克隆转染方法,能使转染后的细胞株稳定或瞬时表达CD36蛋白。
目前报道的导致CD36缺失的CD36基因突变的研究多停留在CD36 cDNA测序及原核克隆,未见建立导致人类CD36缺失的CD36突变基因真核稳定表达细胞株的方法和相关专利的报道。建立导致人类CD36缺失的CD36突变基因真核稳定表达细胞株,才能为研究CD36的结构和功能、阐明所参与的生理和病理机制、探讨在临床治疗中角色和相关药物研发等工作,提高的十分关键的工作资源和平台。
为此本申请人研发了CD36突变基因真核稳定表达细胞株的建立方法,旨在应用所研发的细胞工程技术,建立导致人类CD36缺失的CD36突变基因真核稳定表达细胞株,同时也适用于正常的CD36基因真核稳定表达细胞株,以此解决CD36相关生理、病理、功能、药物等研究以及临床应用的关键问题。
技术实现要素:
一,本发明目的:基于CD36基因遗传学和CD36对人类相关生理、病理、功能和临床
医学的重要作用的深入了解,认识到导致人类CD36缺乏的CD36突变基因【如CD36 C220T(GenBank号:KF539919.1)、329-330del.AC等等】资源,是对CD36研究和应用的十分重要的关键因素。本发明的目的是研发CD36突变基因真核稳定表达细胞株的建立方法,同时该方法也适用于建立正常CD36基因真核稳定表达细胞株。该方法的建立,不仅能解决各类CD36突变基因和表达蛋白的永久稳定保存问题,为CD36的分子结构和功能研究,以及对参与人类体内不同生理和病理过程机制的阐明和临床医学的重要作用的深入探讨,提供了可无限提供的,极其珍贵的遗传资源,同时为与CD36相关的止血、血栓、血小板同种免疫性疾病、高胆固醇血症、肥胖症、外周动脉粥样硬化、动脉高血压、心肌病、糖尿病、疟疾、早老年型痴呆(阿尔茨海默病)等疾病的药物研发和药物靶点的筛选,提供优质的工作平台;特别是能直接用作为检测导致血小板输注无效症的抗-CD36抗体的筛查谱细胞,确保血小板输血治疗的安全和有效性。本研发的方法,将会体现重大的社会和经济价值。
二,本发明的方法步骤:
为实现上述目的,本发明提供一种CD36突变基因真核稳定表达细胞株的建立方法,该方法包括:
① 首先从CD36缺失个体全血标本提取总RNA,由一对PCR扩增引物对CD36 mRNA蛋白编码区进行反转录PCR扩增,获取导致CD36缺失的CD36突变基因 cDNA序列片段,所述的反转录PCR扩增引物包括上游引物YC-36F和下游引物YC-36R,其中,上游引物YC-36F的序列为:5’-ATC CTC GAG ATG GGC TGT GAC CGG A-3’,下游引物YC-36R的序列为:5’-GCA GAA TTC GTT TTA TTG TTT TCG ATC-3’;
②通过四条正向和反向引物,使用重叠延伸PCR技术,该技术英文全称为gene splicing by overlap extension PCR,简称SOE-PCR,拼接扩增CD36突变基因和EGFP荧光基因,获取包含CD36突变基因cDNA编码区全段、EGFP荧光基因全段和所需酶切位点的突变基因序列片段MT-CD36-EGFP;所述SOE-PCR引物包括:
扩增获取包含EcoR 1 酶切位点及保护碱基、CD36蛋白编码区全段的基因序列、融合蛋白基因BamH I及保护碱基、荧光报告基因EGFP首端部分序列的目的基因片段MT-CD36-EGFP-1的正向引物CD36-F2和反向引物CD36-R3,且正向引物CD36-F2亦为后续SOE-PCR拼接扩增MT-CD36-EGFP的正向引物,其序列为:5’-CCG GAA TTC ATG GGC TGT GAC CGG AAC T-3’,反向引物CD36-R3为:5’-TGC TCA CCA TGG ATC CGC GTT TTA TTG TTT TCG ATC TGC-3’;
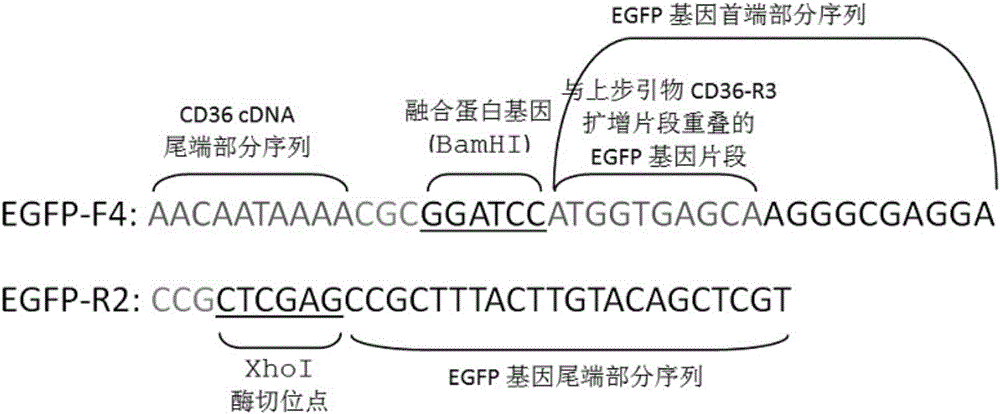
扩增获取包含部分CD36 编码区cDNA片段尾端片段、融合蛋白基因BamH I及保护碱基、荧光报告基因EGFP全段的基因序列、XhoI酶切位点及保护碱基的目的基因片段MT-CD36-EGFP-2的正向引物EGFP-F4和反向引物EGFP-R2,且该反向引物EGFP-R2亦为后续SOE-PCR拼接扩增MT-CD36-EGFP的反向引物,其正向引物EGFP-F4的序列为:5’-AAC AAT AAA ACG CGG ATC CAT GGT GAG CAA GGG CGA GGA-3’,反向引物EGFP-R2的序列为:5’-CCG CTC GAG CCG CTT TAC TTG TAC AGC TCG T-3’;
③将MT-CD36-EGFP连接至pLV4/ StripII-HIS10载体,构建和扩增制备包含CD36突变基因和EGFP荧光基因的MT-CD36-EGFP-pLV4/StripII-HIS10真核表达载体;
④应用病毒介导的真核转染技术,将成功构建的MT-CD36-EGFP-pLV4/StripII-HIS10真核表达载体,转染入CHO-K1细胞系,筛选和建立导致CD36缺失的CD36基因突变真核稳定表达细胞株MT-CD36-CHO-K1;
⑤应用所发明的相同方法,建立正常CD36基因真核稳定表达细胞株Normal-CD36-CHO-K1作为实验的阳性对照,建立单纯稳定表达EGFP荧光蛋白的EGFP-pLV4-CHO-K1株作为实验的阴性对照;在阴性对照EGFP-pLV4-CHO-K1株的构建过程中,首先由1对PCR引物扩增获取包含EcoR 1酶切位点及保护碱基、荧光报告基因EGFP全段的基因序列、XhoI酶切位点及保护碱基的目的基因片段EGFP,所述PCR引物包括上游引物EGFP-F3和下游引物EGFP-R2,其上游EGFP-F3的序列为5’-CCG GAA TTC ATG GTG AGC AAG GGC GAG GA-3’,下游引物EGFP-R2既是本权利要求中②所描述的MT-CD36-EGFP-2片段的反向引物。
三,本发明方法的具体说明:
1,步骤①中,反转录扩增CD36突变基因 cDNA(MT-CD36 cDNA)的引物包括上游引物YC-36F和下游引物YC-36R,其中上游引物YC-36F序列为:5’-ATCCTCGAGATGGGCTGTGACCGGA-3’,下游引物YC-36R序列为:5’-GCAGAATTCGTTTTATTGTTTTCGATC-3’。 如果所扩增的目的基因是CD36突变基因属于点突变基因型,则扩增片段长度为1432bp;如为碱基插入或缺失的突变基因,则扩增片段长度按所插入或缺失的碱基数,在1432bp的基础上做相应的增减。使用TaKaRa One Step RNA PCR Kit (AMV)试剂盒(一步法)进行反转录扩增,反应系组成为(总反应体系体积50μl):
10× one step RNA PCR Buffer 5μl
25mM MgCl2 10μl
10mM dNTPs 5μl
Rnase Inhibitor(40u/μl) 1μl
AMV Rtase XL 1μl
AMV-Optimized Taq 1μl
引物YC-36F (20μM) 1μl
引物YC-36R (20μM) 1μl
RNA(100-500ng/μl) 10μl
H2O 15μl
循环参数为:
50°C 30min
94°C 2min
以下条件扩增30个循环:
94°C 30sec
60°C 30sec
72°C 2min
最后以下条件延伸和保存:
72°C 10min
12°C ∞
测序检测MT-CD36 cDNA目的片段序列。
2,步骤②中,所述的SOE-PCR引物包括:
扩增获取包含EcoR 1 酶切位点及保护碱基、CD36蛋白编码区全段的基因序列、融合蛋白基因BamH I及保护碱基、荧光报告基因EGFP首端部分序列的目的基因片段MT-CD36-EGFP-1的正向引物CD36-F2和反向引物CD36-R3,其正向引物CD36-F2的序列为: 5’-CCGGAATTCATGGGCTGTGACCGGAACT-3’,反向引物CD36-R3的序列为:5’-TGCTCACCATGGATCCGCGTTTTATTGTTTTCGATCTGC-3’,引物示意图见图1。扩增目的片段大小,如果所扩增的目的基因是CD36突变基因属于点突变基因型,则扩增片段长度为1441bp;如为碱基插入或缺失的突变基因,则扩增片段长度按所插入或缺失的碱基数,在1441bp的基础上做相应的增减。
扩增获取包含部分CD36 编码区cDNA片段尾端片段、融合蛋白基因BamH I及保护碱基、荧光报告基因EGFP全段的基因序列、XhoI酶切位点及保护碱基的目的基因片段MT-CD36-EGFP-2的正向引物EGFP-F4和反向引物EGFP-R2,其中,正向引物EGFP-F4的序列为:5’-AACAATAAAACGCGGATCCATGGTGAGCAAGGGCGAGGA-3’, 反向引物EGFP-R2的序列为: 5’-CCGCTCGAGCCGCTTTACTTGTACAGCTCGT-3’,引物示意图见图2。扩增目的片段大小为753bp。
以步骤①中获取的MT-CD36 cDNA为模板,以上描述的CD36-F2和CD36-R3分别为正向和反向引物,进行PCR扩增获取MT-CD36-EGFP-1片段,反应体系组成为(总反应体系体积为25μl):
ddH2O 15μl
5×SF Buffer(with 10mM Mgcl2)(Vazyme) 5μl
dNTP mix (10 mM each) 0.5μl
CD36-F2 (10μM) 1μl
CD36-R3(10μM) 1μl
MT-CD36 cDNA 1μl
DMSO 1μl
HS suffer-Fieviy DNA polymerase (Vazyme) 0.5μl
以含荧光报告基因EGFP的真核质粒pEGFP-N1(苏州吉玛基因药物科技有限公司)为模板,以上述描述的EGFP-F4和EGFP-R2为正向和反向引物,进行PCR扩增获取MT-CD36-EGFP-2片段,反应体系组成为(总反应体系体积为25μl):
ddH2O 16μl
5×SF Buffer(with 10mM Mgcl2)(Vazyme) 5μl
dNTP mix (10 mM each) 0.5μl
EGFP-F4 (10μM) 0.5μl
EGFP-R2 (10μM) 0.5μl
pEGFP-N1质粒 1μl
DMSO 1μl
HS suffer-Fieviy DNA polymerase (Vazyme) 0.5μl
MT-CD36-EGFP-1和MT-CD36-EGFP-2片段PCR扩增循环参数均为:
96°C 5min
以下条件扩增33个循环:
96°C 30sec
60°C 30sec
72°C 90sec
最后以下条件延伸和保存:
72°C 10min
12°C ∞
以上述PCR扩增获取的MT-CD36-EGFP-1和MT-CD36-EGFP-2做为模板,以上述的CD36-F2和EGFP-R2为正向和反向引物,使用SOE-PCR拼接扩增CD36突变基因和EGFP荧光基因,获取包含CD36突变基因cDNA编码区全段、融合蛋白基因BamH I及保护碱基、EGFP荧光基因全段和所需EcoR1和XhoI酶切位点的基因序列片段MT-CD36-EGFP,所扩增目的片段大小:如果所扩增的目的基因是CD36突变基因属于点突变基因型,则扩增片段长度为2165bp;如为碱基插入或缺失的突变基因,则扩增片段长度按所插入或缺失的碱基数,在2165bp的基础上做相应的增减。反应体系为:
H2O 24μl
5×SF Buffer(with10mM Mgcl2)(phantaTM) 10μl
dNTP Mix(10mM each) 1μl
MT-CD36-EGFP-1 4μl
MT-CD36-EGFP-2 4μl
phantaTM HS Suffer-Fideliy DNA Polymerase 1μl
循环参数为:
98°C 5min
以下条件扩增15个循环:
98°C 5min
72°C 1min
待上述反应结束后在反应管中加入以下体系:
CD36-F2(10μM) 2μl
EGFP-R2(10μM) 2μl
DMSO 2μl
再以以下循环参数继续扩增:
以下条件扩增20个循环:
98°C 10sec
56°C 30sec
72°C 60sec
最后以下条件延伸和保存:
72°C 10min
12°C ∞
将以上SOE-PCR扩增得到的MT-CD36-EGFP产物(示意图见图3),进行1.5%琼脂糖凝胶电泳(105v,40min)、切胶,使用AxyPrep DNA Gel Extraction Kit(美国,AXYGEN)胶回收,将使用胶回收产物进行测序检测,验证扩增产物与目的片段序列一致后,该胶回收产物可应用于步骤③的真核表达载体构建工作。
3,步骤③中,首先使用DNA内切酶(EcoR1/Xhol)试剂盒(碧云天),分别对步骤②最后获取的经胶回收纯化的目的基因片段MT-CD36-EGFP和真核载体pLV4/ StripII-HIS10(深圳盎然生物技术有限公司)进行双酶切,反应体系为(总反应体系体积为20μl):
MT-CD36-EGFP或真核载体pLV4/ StripII-HIS10 14μl
10×Buffer Y 2μl
ECOR 1 1μl
Xhol 1μl
ddH2O 2μl
酶切反应条件为:37℃孵育1小时。
酶切反应结束后,对酶切产物进行纯化,纯化方法为:
在酶切产物中加入2-3倍体积的预先冻存于-80℃的无水乙醇,于-20℃静置20min,4℃离心10min,去尽上清,晾干乙醇(5-8min),加入12μl TE Buffer(Elution Buffer)后静置8min,分别测MT-CD36-EGFP及真核载体pLV4/ StripII-HIS10的浓度后,于-20℃保存待用。
随后,将纯化后的MT-CD36-EGFP和真核载体pLV4/ StripII-HIS10酶切产物,使用T4 DNA连接酶试剂盒(Promega公司)进行连接,以获取包含目的CD36突变基因片段和EGFP荧光基因的pLV4/StripII-HIS10表达载体(以下称为MT-CD36-EGFP-pLV4/StripII-HIS10),连接反应体系为:
MT-CD36-EGFP :真核载体pLV4/ StripII-HIS10 摩尔比为7:1(总体积1-8μl)
T4 DAN ligase 10×Buffer 1μl
T4 DAN ligase 1μl
加H2O至终体积为10μl。
连接反应条件:4℃过夜。
最后,将以上连接产物,转化入DH5α Chemically Competent Cell感受态细胞(北京全式金生物技术(TransGen Biotech)有限公司),挑取阳性克隆的DH5α大肠杆菌细胞株,进行培养扩增及验证和获取构建成功的MT-CD36-EGFP-pLV4/StripII-HIS10真核表达载体,具体方法为:
-80℃冰箱取出DH5α Chemically Competent Cell感受态细胞(北京全式金生物技术(TransGen Biotech)有限公司),冰上放置5min融化。吸取50μl感受态细胞加到10μl的上一步骤的连接产物中,冰浴30min后,于42℃热击1min,冰浴2min,然后加入1ml的SOC培养基,180rpm摇床摇1h。吸取300μl的菌液涂板(已加入氨苄青霉素(Amp,终浓度100ug/ml)的LB培养基平板)。将涂好的平板放至37℃培养箱中培养过夜,从转化长好的平板上挑取单个菌落至1.5ml的离心管中(已加入1ml含Amp的SOC培养基),将挑菌的竹签一一对应转移至0.2ml 离心管中(已加入12μl灭菌ddH2O),做好标记,将1.5ml离心管的菌液37℃摇床摇7-8h。
在培养扩增的同时,使用2×Taq Master Mix试剂盒(Vazyme公司),将以上0.2ml 离心管的菌液于100℃孵育20分钟将菌液裂解后,用于菌落PCR,菌落PCR反应体系(总体积25μl)为:
H2O 0.5μl
2×Taq Master Mix 12.5μl
上游引物CD36-F2(10μM) 1μl
下游引物EGFP-R2(10μM) 1μl
菌液 10μl
循环参数为:
98°C 5min
以下条件扩增33个循环:
98°C 10sec
56°C 30sec
72°C 90sec
最后以下条件延伸和保存:
72°C 10min
12°C ∞
选取菌落PCR证实有目的条带的扩增培养对应菌落,吸取部分进行测序验证,剩余部分保存待用。吸取测序检测验证准确的克隆转化的菌液100 ~200μl加入16ml LB培养液中,于37℃摇床过夜;使用无内毒素质粒小量中抽试剂盒(天根生化科技(北京)有限公司)提取质粒,将抽提好的质粒取5μl进行1.5%琼脂糖凝胶电泳,观察条带大小正确后,剩余部分质粒于-20℃保存待用。
4,步骤④中描述的实验方法包括以下内容:
使用Lenti-PacTM HIV Expresslon packaging Kit试剂盒(美国,GeneCopoeia公司)进行慢病毒包装:选取对数生长期的293T细胞以2×108个传代接种于25ml培养瓶中培养24小时,待细胞密度达70%~80%时用于转染,转染前2 h 将细胞培养基更换为Opti-MEM® I培养基(GIBCO),配制慢病毒包装质粒混合液200μl(在无菌1.5ml离心管中,加入步骤③中构建的MT-CD36-EGFP-pLV4/StripII-HIS10慢病毒重组表达质粒5μg和LentiPac HIV 混合试剂5μl,以Opti-MEM® I培养基稀释至终体积200μl);在另一无菌1.5ml离心管内配制EndoFectin 转染试剂应用液200μl(加入185μl Opti-MEM® I培养基和15μl EndoFectin转染试剂);将配制好的EndoFectin 转染试剂应用液缓慢滴加至慢病毒包装质粒混合液中,边滴加边轻柔摇晃混匀;添加混匀完毕后,于室温孵育溶液10-25 分钟,使DNA-EndoFectin 混合物产生;将DNA-EndoFectin 混合液转移至293T细胞的培养液中,混匀,于5% CO2,37 ℃细胞培养箱中培养8 h 后倒去含有转染混和物的培养基,每瓶细胞加入5ml的含10%胎牛血清的DMEM培养基,再加入10μl的TiterBoost 试剂,轻轻晃动培养瓶,使培养基和试剂混匀,置于37℃, 5% CO2细胞培养箱中培养。
收获病毒液,收集转染后 48 小时和72小时的293T 细胞上清液(48小时收集病毒液后,在培养瓶中添加5ml的含10%胎牛血清的DMEM培养基和10μl的TiterBoost 试剂),于 4 ℃,4000g 离心10 min,除去细胞碎片;以 0.45μm 滤器过滤上清液于15 ml离心管中;进行病毒生物学滴度测定后于-80℃保存待用。
使用病毒介导转染建立稳定表达CD36突变基因的CHO-K1细胞株(MT-CD36-CHO-K1株)。取对数生长期的CHO-K1细胞4×107个接种于25ml培养瓶中,加入含10%胎牛血清和1%青霉素-链霉素双抗溶液的DMEM培养基培养24小时,待细胞密度达80%~90%时,弃去旧的培养基,加入4 ml 的DMEM 培养基(含10%热灭活胎牛血清和1%青霉素-链霉素双抗溶液)和100μl病毒液(病毒滴度2×105TU/ml),于5%CO2 ,37℃条件下培养过夜后,弃去原培养基,使用5ml含0.2ug/ml嘌呤霉素(Sigma)的DMEM 培养基(含10%热灭活胎牛血清和1%青霉素-链霉素双抗溶液)进行细胞筛选,此时成功转染入慢病毒包装质粒的细胞在荧光显微镜下观察呈现绿色荧光,而未成功转染的细胞无荧光。加入含嘌呤霉素的培养基筛选后,每24小时观察细胞状态以此,每隔48小时更换含嘌呤霉素的培养基继续筛选培养,筛选培养至少一个月。
对所建立的MT-CD36-CHO-K1株进行验证:收集对数生长期的MT-CD36-CHO-K1株,提取RNA,按步骤①方法进行反转录PCR扩增,获取CD36突变基因 cDNA序列片段并进行测序验证,测序结果与步骤①测序结果一致,表示所建立的MT-CD36-CHO-K1株在分子水平能准确表达CD36突变基因;使用流式细胞术验证MT-CD36-CHO-K1株的细胞表面CD36表达情况;使用蛋白印记实验(western-blotting)验证MT-CD36-CHO-K1株CD36蛋白表达情况;
5,步骤⑤,所述实验阳性对照稳定表达正常CD36的Normal-CD36-CHO-K1株和阴性对照单纯稳定表达EGFP荧光蛋白的EGFP-pLV4-CHO-K1株的建立方法为:
本发明的从CD36表达阳性个体全血标本提取总RNA,按步骤①~④方法,同时建立稳定表达正常CD36基因的Normal-CD36-CHO-K1株作为实验阳性对照,该细胞株CD36表达阳性,在蛋白印记实验(western-blotting)中,所表达的CD36-EGFP融合蛋白可能由于蛋白的不同构象原因,在115000D大小位置附近出现两条CD36阳性蛋白印记条带。
建立转入EGFP荧光基因质粒的单纯稳定表达EGFP荧光蛋白的EGFP-pLV4-CHO-K1株作为实验阴性对照。实验方法为:
扩增获取包含EcoR 1酶切位点及保护碱基 、荧光报告基因EGFP全段的基因序列、XhoI酶切位点及保护碱基的目的基因片段EGFP的正向和反向引物,其正向引物EGFP-F3的序列为:5’-CCGGAATTCATGGTGAGCAAGGGCGAGGA-3’,反向引物使用步骤②中MT-CD36-EGFP-2片段扩增的下游引物EGFP-R2。扩增目的片段大小为743bp。
以含有荧光报告基因EGFP的真核质粒pEGFP-N1(苏州吉玛基因药物科技有限公司)为模板,以上述描述的EGFP-F3和EGFP-R2为正向和反向引物,进行PCR扩增获取EGFP片段,反应体系组成为(总反应体系体积为25μl):
ddH2O 16μl
5×SF Buffer(with 10mM Mgcl2)(Vazyme) 5μl
dNTP mix (10 mM each) 0.5μl
EGFP-F3 (10μM) 0.5μl
EGFP-R2 (10μM) 0.5μl
pEGFP-N1质粒 1μl
DMSO 1μl
HS suffer-Fieviy DNA polymerase (Vazyme) 0.5μl
PCR扩增循环参数与步骤②中MT-CD36-EGFP-1和MT-CD36-EGFP-2扩增参数相同。
将扩增得到的EGFP产物,进行1.5%琼脂糖凝胶电泳(105v,40min)、切胶、使用AxyPrep DNA Gel Extraction Kit(美国,AXYGEN)胶回收,使用胶回收产物进行测序检测,验证扩增产物与目的片段序列一致后,按步骤③和④进行EGFP真核表达载体构建和建立单纯稳定表达EGFP荧光蛋白的EGFP-pLV4-CHO-K1株。
四,发明的重要方法特点和贡献
本发明是基于CD36基因遗传学和CD36对人类相关生理、病理、功能和临床医学的重要作用的深入了解,认识到导致人类CD36缺乏的CD36突变基因资源,是对CD36研究和应用的十分重要的关键因素。研发了CD36突变基因真核稳定表达细胞株的建立方法,同时该方法也适用于建立正常CD36基因真核稳定表达细胞株。
本发明的方法中,通过两条mRNA反转录PCR引物和四条重叠延伸PCR(SOE-PCR)引物,以及探索最佳退火温度及调节Mg2+离子和引物浓度等条件为CD36突变基因真核载体的构建准备接入的目的基因片段;通过探索CD36突变基因真核载体构建的最佳酶切反应条件、插入片段与真核载体连接的反应条件等构建CD36突变基因真核载体;通过探索利用病毒介导转染法建立稳定表达CD36突变基因的真核表达细胞株的病毒包装、转染、药物筛选等条件,达到了建立导致人类CD36缺失的CD36突变基因真核稳定表达细胞株的目的。由于设计引物时充分考虑了CD36开放阅读框基因片段的完整性及EGFP荧光基因的表达,确保了接入目的CD36基因片段和EGFP荧光基因的完整性,同时接入的EGFP荧光基因使得后续建立真核稳定表达细胞株的筛选过程中,得以通过在荧光显微镜下观察细胞筛选情况。在导致人类CD36缺失的CD36突变基因真核稳定表达细胞株建立的过程中,反转录PCR、SOE-PCR和构建的MT-CD36-EGFP-pLV4/StripII-HIS10真核表达载体等步骤均采用基因测序的方式确保了每一步骤目的片段序列的准确性,对所建立的CD36突变基因真核稳定表达CHO-K1细胞株(MT-CD36-CHO-K1株),通过提取RNA进行反转录后测序的方式在分子水平验证了所接入目的MT-CD36基因在真核表达细胞株的表达情况;通过流式细胞术和蛋白印记实验(western-blotting)观察验证MT-CD36基因所造成的CD36蛋白表达变化情况。该方法不仅适合于导致CD36缺失的CD36基因突变如单核苷酸替代、碱基插入、碱基缺失等(但突变点不能位于蛋白表达启动子ATG始 5’至3’方向和终止密码子始3’至5’方向各18个碱基部位)的真核稳定表达细胞株的建立,而且也适用于CD36阳性的正常CD36基因真核稳定表达细胞株的建立。
该方法的建立的贡献,将简单归纳为以下主要方面:
1,所研发的CD36突变基因真核稳定表达细胞株的建立方法,也适用于建立正常CD36基因真核稳定表达细胞株;
2,解决各类CD36突变基因和表达蛋白的永久稳定保存和制备问题;为研究CD36的分子结构、表达、调控和功能,阐明CD36参与人类体内不同生理和病理过程机制和深入探讨在临床医学治疗的重要角色,提供了可无限量的珍贵的遗传资源基础;
3,为与CD36相关的止血、血栓、血小板同种免疫性疾病、高胆固醇血症、肥胖症、外周动脉粥样硬化、动脉高血压、心肌病、糖尿病、疟疾、早老年型痴呆(阿尔茨海默病)等疾病的药物研发和药物靶点的筛选,提供优质的工作平台;
4,能直接用作为检测导致输血小板无效症的抗-CD36抗体的筛查谱细胞,确保血小板输血治疗的安全和有效性。
附图说明
图1为SOE-PCR 中CD36-EGFP-1目的片段扩增引物示意图;
图2为SOE-PCR 中CD36-EGFP-2目的片段扩增引物示意图;
图3为SOE-PCR最终目的片段MT-CD36-EGFP示意图;
图4为实施例1 CD36缺失个体ZYT CD36外显子测序结果图(测序结果为:exon-4具有C220T突变杂合子);
图5 为实施例1 CD36缺失个体ZYT-CD36 cDNA(①)、目的片段ZYT-CD36-EGFP-1(②)和ZYT-CD36-EGFP-2(③)、SOE-PCR连接产物ZYT-CD36-EGFP(④)扩增反应效果图;
图6为实施例1 CD36缺失个体ZYT CD36 cDNA测序结果图(cDNA测序结果显示为220C>T(Gln74Stop)突变)
图7为实施例1成功构建的220T-CD36-EGFP-pLV4/ StripII-HIS10真核表达载体部分测序结果图
图8为实施例1 CD36突变基因(220C>T)真核稳定表达CHO-K1细胞株(220T-CD36-CHO-K1株)cDNA测序验证结果图
图9为实施例1 CD36突变基因(220C>T)真核稳定表达CHO-K1细胞株(220T-CD36-CHO-K1株)CD36蛋白表达流式细胞术实验验证结果图(①:220T-CD36-CHO-K1株CD36蛋白表达阴性;②:阴性对照EGFP-pLV4-CHO-K1株CD36蛋白表达为阴性;③空白对照:CHO-K1株CD36蛋白表达为阴性;④:阳性对照:NORMAL-CD36-CHO-K1株CD36蛋白表达为阳性。)
图10为实施例1 CD36突变基因(220C>T)真核稳定表达CHO-K1细胞株(220T-CD36-CHO-K1株)CD36蛋白表达蛋白印记实验验证结果图(①:220T-CD36-CHO-K1株CD36蛋白表达阴性;②:阴性对照EGFP-pLV4-CHO-K1株CD36蛋白表达为阴性;③空白对照:CHO-K1株CD36蛋白表达为阴性;④:阳性对照:NORMAL-CD36-CHO-K1株CD36蛋白表达为阳性。)。
具体实施方式
下列实施例是对本发明的进一步解释和说明,对本发明不构成任何限制。
本发明是基于导致CD36缺失的分子遗传基础,提供了一种导致人类CD36缺失的CD36突变基因真核稳定表达细胞株建立的方法,通过设计CD36突变基因的反转录引物和真核载体构建引物、探索最佳退火温度及调节Mg2+离子和引物浓度等条件,探索CD36突变基因真核载体构建的最佳酶切反应条件、插入片段与真核载体连接的反应条件等,及探索利用病毒介导转染法建立稳定表达CD36突变基因的真核表达细胞株的病毒包装、转染、药物筛选等条件,可以建立导致人类CD36缺失的CD36突变基因真核稳定表达细胞株。
实施例1
本实例具体介绍通过本发明的CD36突变基因真核稳定表达细胞株的建立方法,建立的导致CD36缺失的220C>T(Gln74stop)(GenBank号:KF539919.1)CD36突变基因的真核稳定表达细胞株,“220T-CD36-CHO-K1株”的实施方案。
首先选择已进行CD36外显子测序确认具有C220T基因突变的CD36缺失个体(该个体标记为ZYT,外显子测序结果见图4),在其的知情同意下抽取静脉血5ml,EDTA抗凝,提取该血液样本总RNA,取本发明第一对PCR引物的上游引物YC-36F和下游引物YC-36R,使用TaKaRa One Step RNA PCR Kit (AMV)试剂盒反转录PCR扩增CD36 cDNA目的片段(ZYT-CD36 cDNA),扩增于ABI 9700型PCR仪进行,扩增反应体系为:
10× one step RNA PCR Buffer 5μl
25mM MgCl2 10μl
10mM dNTPs 5μl
Rnase Inhibitor(40u/μl) 1μl
AMV Rtase XL 1μl
AMV-Optimized Taq 1μl
引物YC-36F (20μM) 1μl
引物YC-36R (20μM) 1μl
RNA(100-500ng/μl) 10μl
H2O 15μl
循环参数为:
50°C 30min
94°C 2min
以下条件扩增30个循环:
94°C 30sec
60°C 30sec
72°C 2min
最后以下条件延伸和保存:
72°C 10min
12°C ∞
取5μl PCR产物,经绿如蓝荧光染料(北京天恩泽基因科技有限公司)染色,于1.5%琼脂糖凝胶中电泳;对照Vazyme公司DL2000 Plus DNAMarker分子标记,在凝胶成像系统中观察特异性PCR产物条带。PCR产物条带清晰、特异,见图5①,取5μl PCR产物进行测序,测序结果见图6,剩余PCR产物于-80℃保存待用。
然后,取本发明中的第二对引物的上游引物CD36-F2和下游引物CD36-R3,以ZYT-CD36 cDNA为DNA模板,扩增含EcoR 1 酶切位点及保护碱基、CD36蛋白编码区全段的基因序列、融合蛋白基因BamH I及保护碱基 、荧光报告基因EGFP首端部分序列的目的基因片段ZYT-CD36-EGFP-1,扩增于ABI 9700型PCR仪进行,扩增反应体系组成为:
ddH2O 15μl
5×SF Buffer(with 10mM Mgcl2)(Vazyme) 5μl
dNTP mix (10 mM each) 0.5μl
CD36-F2 (10μM) 1μl
CD36-R3(10μM) 1μl
ZYT-CD36 cDNA 1μl
DMSO 1μl
HS suffer-Fieviy DNA polymerase (Vazyme) 0.5μl
循环参数为:
96°C 5min
以下条件扩增33个循环:
96°C 30sec
60°C 30sec
72°C 90sec
最后以下条件延伸和保存:
72°C 10min
12°C ∞
取5μl PCR产物,经绿如蓝荧光染料(北京天恩泽基因科技有限公司)染色,于1.5%琼脂糖凝胶中电泳;对照Vazyme公司DL2000 Plus DNAMarker分子标记,在凝胶成像系统中观察特异性PCR产物条带。PCR产物条带清晰、特异,见图5②,剩余PCR产物于-80℃保存待用。
取本发明中的第三对引物的上游引物EGFP-F4和下游引物EGFP-R2,以苏州吉玛基因药物科技有限公司生产的含有荧光报告基因EGFP的真核质粒pEGFP-N1为模板,扩增获取包含部分CD36 编码区cDNA片段尾端片段、融合蛋白基因BamH I及保护碱基、荧光报告基因EGFP全段的基因序列、XhoI酶切位点及保护碱基的目的基因片段ZYT-CD36-EGFP-2,扩增于ABI 9700型PCR仪进行,扩增反应体系组成为:
ddH2O 16μl
5×SF Buffer(with 10mM Mgcl2)(Vazyme) 5μl
dNTP mix (10 mM each) 0.5μl
EGFP-F4 (10μM) 0.5μl
EGFP-R2 (10μM) 0.5μl
pEGFP-N1质粒 1μl
DMSO 1μl
HS suffer-Fieviy DNA polymerase(Vazyme) 0.5μl
循环参数为:
96°C 5min
以下条件扩增33个循环:
96°C 30sec
60°C 30sec
72°C 90sec
最后以下条件延伸和保存:
72°C 10min
12°C ∞
取5μl PCR产物,经绿如蓝荧光染料(北京天恩泽基因科技有限公司)染色,于1.5%琼脂糖凝胶中电泳;对照Vazyme公司DL2000 Plus DNAMarker分子标记,在凝胶成像系统中观察特异性PCR产物条带。PCR产物条带清晰、特异,图5③,剩余PCR产物于-80℃保存待用。
取本发明中的第二对引物的上游引物CD36-F2和第三对引物的下游引物EGFP-R2,以ZYT-CD36-EGFP-1和ZYT-CD36-EGFP-2为模板,以SOE-PCR拼接扩增获取包含CD36突变基因 ZYT的cDNA编码区全段、融合蛋白基因BamH I及保护碱基、EGFP荧光基因全段和所需EcoR1和XhoI酶切位点的基因序列片段ZYT-CD36-EGFP,扩增于ABI 9700型PCR仪进行,扩增反应体系组成为:
H2O 24μl
5×SF Buffer(with10mM Mgcl2)(phantaTM) 10μl
dNTP Mix(10mM each) 1μl
ZYT-CD36-EGFP-1 4μl
ZYT-CD36-EGFP-2 4μl
phantaTM HS Suffer-Fideliy DNA Polymerase 1μl
循环参数为:
98°C 5min
以下条件扩增15个循环:
98°C 5min
72°C 1min
待上述反应结束后在反应管中加入以下反应体系:
CD36-F2 2μl
EGFP-R2 2μl
DMSO 2μl
再以以下循环参数继续扩增:
以下条件扩增20个循环:
98°C 10sec
56°C 30sec
72°C 60sec
最后以下条件延伸和保存:
72°C 10min
12°C ∞
取PCR产物,经绿如蓝荧光染料(北京天恩泽基因科技有限公司)染色,于1.5%琼脂糖凝胶中电泳;对照Vazyme公司DL2000 Plus DNA Marker分子标记,观察特异性PCR产物条带。PCR产物条带清晰、特异,图5④,切胶切取2165bp位置目的片段、使用AxyPrep DNA Gel Extraction Kit(美国,AXYGEN)胶回收,取胶回收产物4μl进行测序检测,验证扩增产物与目的片段序列一致后,剩余胶回收产物于-80℃保存待用。
获取纯化的ZYT-CD36-EGFP目的片段后,使用DNA内切酶(Eco1/Xhol)试剂盒(碧云天)分别对ZYT-CD36-EGFP和真核载体pLV4/ StripII-HIS10(深圳盎然生物)进行双酶切,反应体系为
ZYT-CD36-EGFP或真核载体pLV4/StripII-HIS10 14μl
10×Buffer Y 2μl
ECOR 1 1μl
Xhol 1μl
ddH2O 2μl
酶切反应条件为:37℃孵育1小时。
酶切反应结束后,对酶切产物进行纯化,纯化方法为:
在酶切产物中加入2-3倍体积的预先冻存于-80℃的无水乙醇,于-20℃静置20min,4℃离心10min,去尽上清,晾干乙醇(5-8min),加入12μl TE Buffer(Elution Buffer)静置8min,分别测ZYT-CD36-EGFP及真核载体pLV4/StripII-HIS10的浓度后,于-20℃保存待用。
使用Promega公司T4 DNA连接酶试剂盒,将纯化后的ZYT-CD36-EGFP和真核载体pLV4/StripII-HIS10酶切产物进行连接,以获取包含目的CD36突变基因片段和EGFP荧光基因的pLV4/StripII-HIS10表达载体(C220T-CD36-EGFP-pLV4/StripII-HIS10),连接反应体系为:
ZYT-CD36-EGFP :真核载体pLV4/StripII-HIS10 摩尔比为7:1(总体积1-8μl)
T4 DAN ligase 10×Buffer 1μl
T4 DAN ligase 1μl
加H2O至终体积为10μl。
连接反应条件:4℃过夜。
将以上连接产物,转化入DH5α Chemically Competent Cell感受态细胞(北京全式金生物技术(TransGen Biotech)有限公司),挑取阳性克隆的DH5α大肠杆菌细胞株,进行培养扩增及验证和获取构建成功的220T-CD36-EGFP-pLV4/ StripII-HIS10真核表达载体,具体方法为:
-80℃冰箱取出DH5α Chemically Competent Cell感受态细胞(北京全式金生物技术(TransGen Biotech)有限公司),冰上放置5min融化。吸取50μl感受态细胞加到10μl的上一步骤的连接产物(在转化前,在连接产物中加入1μl的solutionIII)中,冰浴30min后,于42℃热击1min,冰浴2min,然后加入1ml的SOC培养基,180rpm摇床摇1h。吸取300μl的菌液涂板(已加入氨苄青霉素(Amp,终浓度100ug/ml)的LB平板)。将涂好的平板放至37℃培养箱中培养过夜,从转化长好的平板上挑取单个菌落至1.5ml的离心管中(已加入1ml含Amp的SOC培养基),将挑菌的竹签一一对应转移至0.2ml 离心管中(已加入12μl灭菌ddH2O),做好标记,将1.5ml离心管的菌液37℃摇床摇7-8h。
在培养扩增的同时,取本发明中的第二对引物的上游引物CD36-F2和第三对引物的下游引物EGFP-R2,以0.2ml 离心管的菌液(于PCR实验前,将该菌液于100℃孵育20分钟将菌液裂解后用于实验)作为模板,使用Vazyme公司2×Taq Master Mix试剂盒进行菌落PCR扩增,扩增于ABI 9700型PCR仪进行,扩增反应体系组成为:
H2O 0.5μl
2×Taq Master Mix 12.5μl
上游引物CD36-F2(10μM) 1μl
下游引物EGFP-R2(10μM) 1μl
菌液 10μl
循环参数为:
98°C 5min
以下条件扩增33个循环:
98°C 10sec
56°C 30sec
72°C 90sec
最后以下条件延伸和保存:
72°C 10min
12°C ∞
取5μl PCR产物,经绿如蓝荧光染料(北京天恩泽基因科技有限公司)染色,于1.5%琼脂糖凝胶中电泳;对照Vazyme公司DL2000 Plus DNA Marker分子标记,在凝胶成像系统中观察特异性PCR产物条带。选取菌落PCR产物条带清晰、特异,片段大小与SOE-PCR扩增片段产物ZYT-CD36-EGFP大小一致扩增培养对应菌落,吸取部分进行测序验证,剩余部分保存待用。吸取测序检测验证结果为CD36 220T(见图7)且其余部分序列与接入ZYT-CD36-EGFP序列一致的克隆转化菌液100 ~200μl加入16ml LB培养液中,于37℃摇床过夜,扩增培养获取构建成功的220T-CD36-EGFP-pLV4/StripII-HIS10真核表达载体;使用无内毒素质粒小量中抽试剂盒(天根生化科技(北京)有限公司)提取质粒,将抽提好的质粒取5μl进行1.5%琼脂糖凝胶电泳,观察条带大小正确后,剩余部分质粒于-20℃保存待用。
最后,使用病毒介导法真核转染技术,将成功构建的220T-CD36-EGFP-pLV4/StripII-HIS10真核表达载体,转染入CHO-K1细胞系,筛选和建立导致CD36缺失的CD36 220C>T突变基因真核稳定表达细胞株(220T-CD36-CHO-K1),具体方法为:
使用美国GeneCopoeia公司Lenti-PacTM HIV Expresslon packaging Kit试剂盒进行慢病毒包装。选取对数生长期的293T细胞以2×108个传代接种于25ml培养瓶中培养24小时,待细胞密度达70%~80%时用于转染,转染前2 h 将细胞培养基更换为GIBCO公司Opti-MEM® I培养基,配制慢病毒包装质粒混合液200μl(在无菌1.5ml离心管中,成功构建的220T-CD36-EGFP-pLV4/StripII-HIS10真核表达质粒5μg和LentiPac HIV混合试剂5μl,以Opti-MEM® I培养基稀释至终体积200μl);在另一无菌1.5ml离心管内配制EndoFectin 转染试剂应用液200μl(加入185μl Opti-MEM® I培养基和15μl EndoFectin转染试剂);将配制好的EndoFectin 转染试剂应用液缓慢滴加至慢病毒包装质粒混合液中,边滴加边轻柔摇晃混匀;添加混匀完毕后,于室温孵育溶液10-25 分钟,使DNA-EndoFectin 混合物产生;将DNA-EndoFectin 混合液转移至293T细胞的培养液中,混匀,于5% CO2,37℃细胞培养箱中培养8 h 后倒去含有转染混和物的培养基,每瓶细胞加入5ml的含10%胎牛血清的DMEM培养基,再加入10μl的TiterBoost 试剂,轻轻晃动培养瓶,使培养基和试剂混匀,置于37℃, 5% CO2细胞培养箱中培养。
收获病毒液:收集转染后48小时和72小时的293T 细胞上清液(48小时收集病毒液后,在培养瓶中添加5ml的含10%胎牛血清的DMEM培养基和10μl的TiterBoost 试剂),于 4 ℃,4000g 离心10 min,除去细胞碎片;以 0.45 μm 滤器过滤上清液于15 ml离心管中;进行病毒生物学滴度测定后于-80℃保存待用。
使用所收获的病毒液介导转染CHO-K1细胞株,并使用嘌呤霉素进行筛选建立CD36 220C>T突变基因真核稳定表达细胞株(220T-CD36-CHO-K1株):取对数生长期的CHO-K1细胞4×107个接种于25ml培养瓶中,加入含10%胎牛血清和1%青霉素-链霉素双抗溶液的DMEM培养基培养24小时,待细胞密度达80%~90%时,弃去旧的培养基,加入4 ml 的DMEM 培养基(含10%热灭活胎牛血清和1%青霉素-链霉素双抗溶液)和100μl病毒液(病毒滴度2×105TU/ml),于5%CO2 ,37℃条件下培养过夜后,弃去原培养基,使用5ml含0.2ug/ml嘌呤霉素(Sigma)的DMEM 培养基(含10%热灭活胎牛血清和1%青霉素-链霉素双抗溶液)的进行细胞筛选,每24小时观察细胞状态,以此,每隔48小时更换含嘌呤霉素的培养基继续筛选培养,筛选培养至少1个月。
同时建立稳定表达正常CD36的Normal-CD36-CHO-K1株作为实验的阳性对照,只转入EGFP荧光基因质粒的单纯稳定表达EGFP荧光蛋白的EGFP-pLV4-CHO-K1株等分别作为实验的阴性对照,方法如下:
从CD36表达阳性个体全血标本提取总RNA,按以上步骤方法,同时建立稳定表达正常CD36的Normal-CD36-CHO-K1株作为实验阳性对照。
建立转入EGFP荧光基因质粒的单纯稳定表达EGFP荧光蛋白的EGFP-pLV4-CHO-K1株作为实验阴性对照。实验方法为:
以含有荧光报告基因EGFP的真核质粒pEGFP-N1(苏州吉玛基因药物科技有限公司)为模板,EGFP-F3和EGFP-R2为正向和反向引物,进行PCR扩增获取包含EcoR 1酶切位点及保护碱基、荧光报告基因EGFP全段的基因序列、XhoI酶切位点及保护碱基的目的基因片段EGFP,反应体系组成为(总反应体系体积25μl):
ddH2O 16μl
5×SF Buffer(with 10mM Mgcl2)(Vazyme) 5μl
dNTP mix (10 mM each) 0.5μl
EGFP-F3 (10μM) 0.5μl
EGFP-R2 (10μM) 0.5μl
pEGFP-N1质粒 1μl
DMSO 1μl
HS suffer-Fieviy DNA polymerase (Vazyme) 0.5μl
PCR扩增循环参数与上述步骤中ZYT-CD36-EGFP-1和ZYT-CD36-EGFP-2目的片段扩增的参数相同。
将扩增得到的EGFP产物,进行1.5%琼脂糖凝胶电泳(105v,40min)、切胶、使用AxyPrep DNA Gel Extraction Kit(美国,AXYGEN)胶回收,使用胶回收产物进行测序检测,验证扩增产物与目的片段序列一致后,按上述220T-CD36-EGFP-pLV4/StripII-HIS10质粒的构建和220T-CD36-CHO-K1细胞株的筛选建立方法,进行EGFP真核表达载体构建和建立单纯稳定表达EGFP荧光蛋白的EGFP-pLV4-CHO-K1株。
对所建立的220T-CD36-CHO-K1株进行验证:
收集对数生长期的220T-CD36-CHO-K1株、阳性对照Normal-CD36-CHO-K1株,分别提取RNA,取本发明第一对PCR引物的上游引物YC-36F和下游引物YC-36R,使用TaKaRa One Step RNA PCR Kit (AMV)试剂盒反转录PCR扩增CD36 cDNA目的片段,扩增于ABI 9700型PCR仪进行,扩增反应体系及循环参数如第一步所述,取5μl PCR产物,经绿如蓝荧光染料(北京天恩泽基因科技有限公司)染色,于1.5%琼脂糖凝胶中电泳;对照VAZYME公司DL2000 PLUS DNA MARKER分子标记,在凝胶成像系统中观察特异性PCR产物条带。PCR产物条带清晰、特异,大小与第一步扩增的ZYT-CD36 cDNA片段大小一致,取5μl PCR产物进行测序,220T-CD36-CHO-K1株cDNA测序结果序列与所转染质粒220T-CD36-EGFP-pLV4/StripII-HIS10中CD36基因片段序列一致,见图8,表示所建立的220T-CD36-CHO-K1株在分子水平能准确表达220C>T CD36突变基因;阳性对照Normal-CD36-CHO-K1株cDNA测序结果序列与所转染质粒Normal-CD36-EGFP-pLV4/ StripII-HIS10中CD36基因片段序列一致,表示所建立的Normal-CD36-CHO-K1株在分子水平能准确表达正常CD36基因;阴性对照EGFP-pLV4-CHO-K1株cDNA测序结果序列与所转染质粒EGFP-pLV4/ StripII-HIS10中EGFP基因片段序列一致,表示所建立的EGFP-pLV4-CHO-K1株在分子水平能准确表达EGFP基因。
收集对数生长期的220T-CD36-CHO-K1株、阳性对照Normal-CD36-CHO-K1株、阴性对照EGFP-pLV4-CHO-K1株、空白对照CHO-K1株,使用流式细胞术检测这几个细胞株CD36表达情况,结果见图9,220T-CD36-CHO-K1株、阴性对照EGFP-pLV4-CHO-K1株、空白对照CHO-K1株检测结果均为CD36表达阴性,阳性对照NORMAL-CD36-CHO-K1株检测结果为CD36表达阳性;分别提取这几个细胞株的膜总蛋白,使用蛋白印记实验(western-blotting)检测各细胞株CD36蛋白表达情况,结果见图10,220T-CD36-CHO-K1株、阴性对照EGFP-pLV4-CHO-K1株、空白对照CHO-K1株检测结果均为CD36表达阴性,阳性对照Normal-CD36-CHO-K1株检测结果为CD36表达阳性。
- 该技术已申请专利。仅供学习研究,如用于商业用途,请联系技术所有人。
- 技术研发人员:吴国光;李丽兰;蒋丽红;黎海燕;陈洁润;
- 技术所有人:南宁输血医学研究所;南宁中心血站;
- 我是此专利的发明人
- 该领域下的技术专家
- 如您需求助技术专家,请点此查看客服电话进行咨询。
- 1、薛老师:1.CRISPR-Cas系统 2.基因编辑 3.基因修复 4.天然产物合成 5.单分子技术开发与应用
- 2、张老师:1.探索新型氧化还原酶结构-功能关系,电催化反应机制 2.酶电催化导向的酶分子改造 3.纳米材料、生物功能多肽对酶-电极体系的影响4. 生物电化学传感和生物电合成体系的设计与应用。
- 3、豆老师:1.环境纳米材料及挥发性有机化合物(VOCs) 2.CO污染物的催化氧化 3.低温等离子体 4.吸脱附等控制技术
- 4、赵老师:1.高分子材料改性及加工技术 2.微孔及过滤材料 3.环境友好高分子材料
- 5、邬老师:1.高分子材料的共混与复合 2.涉及材料功能化及结构与性能的研究; 高分子热稳定剂的研发
- 如您是高校老师,可以点此联系我们加入专家库。
- 还没有人留言评论。精彩留言会获得点赞!