用于基因组DNA片段的靶向纯化的制备型电泳方法与流程
本申请要求2015年11月20日提交的标题为“PREPARATIVE ELECTROPHORETIC METHOD FOR TARGETED PURIFICATION OF GENOMIC DNA FRAGMENTS”的美国临时专利申请NO.62/258,384的优先权,其公开内容以引用的方式整体并入本文中。
介绍
由于全基因组测序的高成本和全基因组序列分析的复杂性,大多数下一代测序实验试图检查基因组的靶向区域,例如所有蛋白质编码区(整个外显子组测序,“WES”)或所谓的靶向测序研究中的特定基因子集(或基因组区域的特定子集)。当使用条形码化的测序适配器进行这样的靶向测序实验时,许多这样的样品可以合并并且在单次测序运行中一起运行,由此显着降低每个样品测序成本。基于这个原因,今天(以及过去几年)进行的绝大多数NGS都是WES或靶向测序。
用于制备靶向测序文库的最常用方法分为两种通用策略1)杂交捕获,或2)扩增子文库构建。
在杂交捕获方法中,靶向基因组DNA片段变性为单链形式并在溶液中与生物素标记的单链核酸探针(也称为“诱饵”)杂交。杂交后,将生物素标记的杂交物捕获到抗生物素蛋白或抗生蛋白链菌素涂覆的微粒(通常顺磁性颗粒)上,然后通过磁性或离心收集颗粒与溶液相非靶向基因组DNA分离。
文库构建可以在靶序列捕获之前或之后进行。在最常用的技术形式(如Agilent Sureselect和Illumina全外显子试剂盒)中,在杂交捕获之前进行DNA片段化,末端修复和测序适配器连接。然而,在一些靶向测序方案中,杂交捕获先于文库构建(Wang等,BMC Genomics,(2015)16:214;DOI 10.1186/s12864-015-1370-2)。
杂交探针可以是单链DNA,RNA或其类似物。许多人发现将捕获探针设计为含有强RNA聚合酶启动子(如T7RNA聚合酶启动子)的DNA寡核苷酸是方便的,从而可以通过体外转录反应廉价地生产生物素化的单链RNA捕获探针。当需要在大量样品中检测相同的靶向组时,这样的方法特别有用(Gnirke等,Nature Biotechnology,(2009)27:182)。
在扩增子文库法中,首先通过PCR扩增用于测序的目标区域,然后将PCR产物用作NGS文库构建的DNA输入。这种方法最流行的例子是Life Technologies公司的AmpliSeq靶向测序试剂盒(举例而言)。扩增子测序方法在临床实验室中很受欢迎,因为PCR允许使用较小的输入样本量(1-10ng输入DNA)。
靶向测序的两种方法都具有共同的缺点,因为它们需要相对大量的核酸试剂:杂交捕获方法中的生物素化探针,或扩增子测序情况下的PCR引物对。当使用普通的短读测序(short-read sequencing)技术如Illumina或Ion Torrent(典型的原始阅读长度<400bp)来检查基因组DNA的长的几千碱基(kb)区域时,这尤其值得关注。一个相关的问题是,在便利的多重形式中设计和优化试剂盒的复杂性,所述试剂盒使用具有所需特异性/严格性的复杂探针或引物组。
目前的靶向测序方法的另一困难是难以将它们应用于非常大的DNA分子(10kb直到低单个兆碱基对(mb)),用于研究长距离基因组结构和重排,以及用于长距离定相(long-range phasing)和单倍型分析。例如,长距离PCR对于大于10kb的距离是不可靠的,并且大多数商业可用的DNA提取方法不能可靠地产生大于50-100kb的DNA。
一些实施方案的概述
在本公开的一些实施方案中,提供了用于分离基因组DNA的特定片段的方法,其包括:
·提供含有高分子量(HMW)DNA的样品;
·将样品截留在凝胶基质中,其中HMW DNA具有极低的电泳迁移率;
·将具有截留的样品的凝胶基质暴露于裂解试剂以从其他样品污染物中释放HMW DNA;
·使具有截留的样品的凝胶基质经受电泳场,使得样品污染物和裂解试剂从凝胶基质中除去,留下凝胶截留的纯化的HMW DNA;
·将具有截留的纯化DNA的凝胶基质用DNA裂解酶试剂处理,该DNA裂解酶试剂被配置为在样品DNA内的特定DNA序列处进行裂解,从而将样品的确定的区段释放为尺寸减小的片段,其中释放的确定的DNA片段具有比未裂解的HMW样品DNA的剩余部分高得多的电泳迁移率;
·使凝胶基质经受电泳场,使得特异性切除的DNA片段与未裂解的HMW样品DNA的剩余部分物理分离;和
·从凝胶基质中分离特异性切除的和电泳分离的DNA片段,留下仍然截留在凝胶基质中的HMW DNA样品的未裂解剩余部分。
在一些实施方案中,提供了包括多个上述步骤的类似方法。
在一些实施方案中,样品包含完整细胞(例如动物,植物,细菌,真菌,古细菌,原生动物)或完整病毒颗粒的液体悬浮液。
在一些实施方案中,凝胶基质包含浓度为约0.2%至约5%(重量/体积)的琼脂糖水凝胶。
在本发明的一些实施方案中,裂解剂包含浓度为0.05%至10%的阴离子去污剂。在本发明的一些实施方案中,阴离子去污剂是十二烷基硫酸钠(SDS)。
在一些实施方案中,通过裂解样品释放的DNA的尺寸是长度大于10兆碱基对,由特异性裂解酶试剂释放的DNA片段的尺寸是长度<2兆碱基对。
在利用细菌、植物或真菌细胞的一些实施方案中,在细胞裂解之前使用另外的酶试剂处理来除去细胞壁。
在一些实施方案中,通过电洗脱将由特异性裂解酶试剂特异性释放的DNA片段从凝胶基质分离至含有液体缓冲液的洗脱模块中。
在一些实施方案中,提供用于截留样品和纯化样品DNA的方法和装置。用于酶处理纯化的截留的样品DNA的一般方法描述于共同未决的PCT申请PCT/US2015/055833,其全部公开内容通过引用整体并入本文。
在一些实施方案中,DNA裂解酶试剂包含一种或多种RNA指导的内切核酸酶组合物。例如,一种这样的组合物基于CRISPR/Cas9系统(参见下文),其包含具有指导RNA的Cas9蛋白,其使得指导RNA-Cas9复合物能够在基因组DNA中的特定的用户指定位点处裂解。
其他序列特异性DNA裂解酶试剂可以与一些实施方案一起使用,诸如其他RNA指导的内切核酸酶系统,工程化的锌指核酸酶,工程化的转录激活物样核酸酶和工程化的大范围核酸酶(Zetsche等,(2015)Cell 163:759;Hsu PD,等,(2014)Cell 157:1262;Esvelt和Wang,(2013)Mol Syst Biol.9:641;Stoddard BL,(2011)Structure 19:7;Urnov FD,等,(2010)Nat Rev Genet.11:636;Bogdanove和Voytas,(2011)Science 333:1843;Scharenberg AM,等,(2013)Curr Gene Ther.13(4):291)。
Cas9蛋白是成簇的规则间隔的短回文重复序列(CRISPR)系统的主要成分,所述系统是细菌中的适应性免疫系统,所述细菌降解入侵病毒的DNA序列(Gasiunas,G.等,(2012)Proc Natl Acad Sci USA,109:E2579;Jinek,M.等,Science,(2012)337:816)。Cas9是具有实现其结合特异性的独特机制的DNA内切核酸酶。Cas9与CRISPR RNA复合,所述CRISPR RNA指导蛋白质结合和裂解与这些RNA序列互补的DNA序列。在内源性CRISPR系统中,多种RNA协同作用以指导Cas9,但是最近已证实Cas9结合由单个嵌合gRNA指导的特定DNA序列。此后,除非另有说明,否则我们将嵌合gRNA简称为gRNA。Cas9的发现催化了将该蛋白质用于基因组编辑的巨大兴趣,因为设计靶向内切核酸酶现在变得像订购寡核苷酸一样简单(Cong,L.等,Science,(2013)339:819;Mali,P.,等,(2013)Science 339:823)。一般通过在细胞中表达Cas9蛋白和指导RNA两者来进行体内的Cas9实现的DNA裂解。然而,体外DNA裂解也可以通过将纯化的Cas9蛋白与gRNA组合并添加DNA模板来实现(Karvelis等,(2013)Biochem Soc Trans 41:1401,PMID 24256227)。实际上,指导RNA-Cas9复合物用作用户可定制的限制性内切酶,允许在几乎任何已知的DNA序列处或附近进行裂解。通过这种方式,用户可以配置CRISPR/Cas9试剂的组合,以裂解样本DNA中感兴趣的特定DNA区域周围的区域。
最常用的Cas9核酸酶来自酿脓链球菌(Streptococcus pyogenes)细菌(SpCas9),并且其在适当条件下、在体外和体内以接近100%的效率特异性裂解DNA(Shalem等,(2014)Science 343:84 PMID:24336571;Wang等,(2014)Science 343:80PMID:24336569)。SpCas9通过RNA中的20nt序列靶向DNA基因座,该序列位于称为原型间隔区邻接基序(protospacer adjacent motif)(PAM)的所需5’NGG基序的直接上游。该20nt gRNA序列将SpCas9蛋白导向其在DNA靶序列中的互补物。该区域中gRNA和靶DNA之间的任何错配都会降低裂解效率。发生在PAM远端的单一错配在某种程度上是可以容忍的(Hsu等,Nat Biotechnol 31:827,PMID:23873081),但是多个错配会导致裂解效率的显着降低。因此,通常需要配置具有靶向序列的gRNA,所述靶向序列与其DNA靶完全互补并且与基因组中的所有其他20聚体具有至少2或3个错配。
为了靶向体外裂解DNA,通常通过将编码所需20nt靶向序列的寡核苷酸克隆到含有T7启动子下游的gRNA恒定区的质粒载体中来制备gRNA。然后可以通过用T7RNA聚合酶进行体外转录反应来将该质粒用于生产gRNA。然后将所得的gRNA纯化并与Cas9蛋白复合。加入靶DNA并在37度下进行裂解反应~1小时。或者,可以通过以下来产生gRNA:合成编码20nt靶向序列和侧翼恒定区的DNA寡核苷酸并进行巨引物(megaprimer)PCR或拼接PCR以添加gRNA的剩余的恒定部分和T7启动子。通常,希望在大量特定基因座处裂解DNA。实现此目的的一种有效方式是在微阵列上合成数百或数千个各自编码不同gRNA靶向序列的DNA寡核苷酸(Singh-Gasson等,(1999)Nat Biotechnol 17:974;Hughes等,(2001)Nat Biotechnol 19:342)。然后可将该文库克隆入含有T7启动子和gRNA恒定区和如上所述产生的gRNA的质粒。或者,可以使用先前描述的基于巨引物或拼接PCR的策略。
附图简述
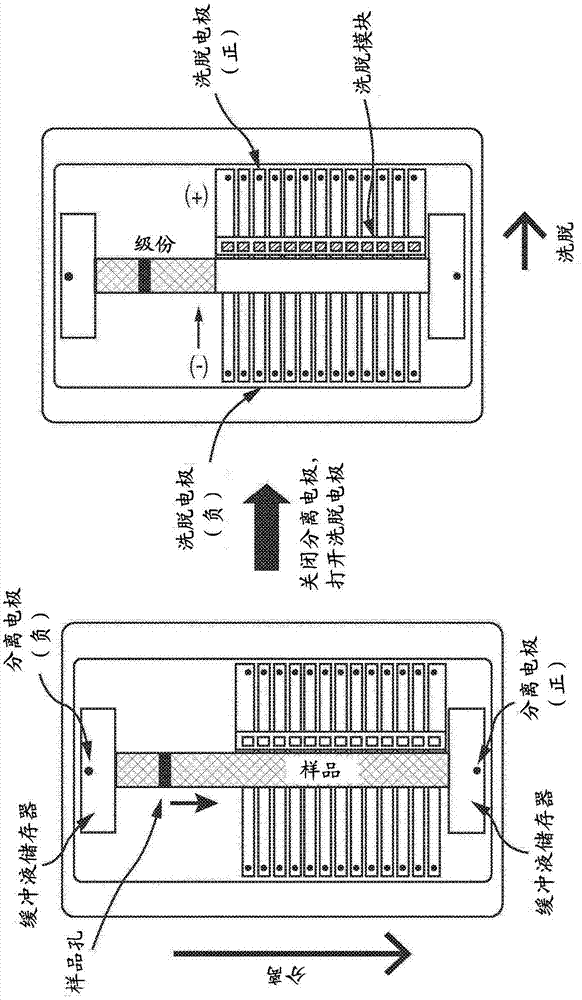
图1示出了美国公开号2015/00101932中描述的制备型电泳盒和尺寸选择过程的示意图,所述美国公开号2015/00101932通过引用整体并入本文。
图2示出了在共同未决的PCT申请PCT/US2015/055833中描述的类型的盒,所述申请在下文中被称为'833PCT,其通过引用整体并入本文。
图3A-E示出了根据本公开的一些实施方案的用于从HMW DNA获得靶向DNA片段的系统和方法的示意图。
图4示出了靶向位点,相关编码区和产生NPM1-ALK融合重排的一些共同断裂点的示意图。
一些实施方案的详细描述
图3A-E示出了利用凝胶电泳盒(例如,如图1和2所示)的本公开内容的实施方案。图1显示了使用该盒的商业制备型电泳盒和尺寸选择过程的示意图。在图1的左侧,样品核酸从样品孔向下电泳通过琼脂糖填充的分离通道,在那里它们按尺寸分开。分离后,关闭分离电极,打开洗脱电极,从而将分离的核酸侧向电洗脱入分离通道右侧的一组洗脱模块。洗脱的核酸在液体电泳缓冲液中,并且可以使用手动或自动移液装置从洗脱模块移除。
如图2所示,图1的盒被修改以提供'833PCT中所述类型的盒。在该盒中,样品孔与样品孔上游的试剂孔(即,对应于负分离电极的近侧的上游)一起被提供。在一些实施方案中,试剂孔的体积比样品孔的体积大,并且比样品孔稍宽且更深以确保样品孔在电泳纯化步骤期间被来自试剂孔的试剂完全包围。
根据本公开内容的一些实施方案,如图3A-E所示的示例性工作流程如下(为了简化说明,在图中仅示出了分离通道和洗脱通道)。图3A显示了刚加载了样品后的盒。优选的是,样品是细胞悬浮液。优选样品的实例可以是全血,纯化的白细胞,培养细胞的悬浮液,微生物悬浮液,从口腔拭子制备的细胞悬浮液,以及从固体组织制备的样品,所述固体组织已经过酶处理以将组织分散为单一细胞悬浮液。优选地,将样品细胞悬浮液装载到等密度加载溶液中,使得细胞在纯化电泳过程中既不会漂浮也不会下沉。
同样在图3A中,裂解试剂被装载在试剂孔中。在一些实施方案中,试剂孔和样品孔在物理上是隔离的并且在加载期间不能混合。在一些实施方案中,裂解试剂包含带负电的组分,其可以电泳通过凝胶基质进入样品孔中,在那里它们将在不混合或搅拌样品孔组分的情况下温和地裂解细胞。以这种方式,裂解发生在无剪切方式中,并且恰恰产生HMW DNA。优选的裂解试剂包括阴离子去污剂如SDS和十二烷基肌氨酸钠(sodium sarkcosyl),以及去污剂耐受性蛋白酶如蛋白酶K,螯合剂如EDTA和EGTA,以及上述物质的混合物。
加载样品孔和试剂孔后,可以使用分离电极施加电泳场。在电泳的初始阶段,将裂解试剂递送至样品孔,在那里细胞在很少或没有粘性剪切流的情况下快速且温和地裂解。在一些实施方案中,蛋白质和其他细胞组分被裂解试剂变性和/或降解,并且被在裂解试剂中的阴离子去污剂负电荷包被。结果,这种污染物从样品孔中很快电泳移动。然而,由于细胞裂解迅速且温和,细胞DNA仍然大部分未降解,并且具有如此大的尺寸(估计大于10兆碱基),因此它不会电泳移动到分离凝胶柱中。纯化电泳的结束如图3B所图示的,其中HMW DNA被捕获在样品孔的下游壁中,并且去污剂胶束和去污剂-蛋白质复合物在凝胶底部。
尽管DNA在该纯化步骤过程中被捕获在凝胶基质中,但是它可以通过如'833PCT中所述的DNA修饰酶如限制性酶,转座酶,聚合酶和外切核酸酶来加工。在图3C中,样品孔和试剂孔被清空,试剂孔重新填充了与CRISPR-Cas9裂解缓冲液相容的缓冲液(参见下面的实例),并且样品孔重新填充了定制CRISPR-Cas9裂解试剂,其被配置为围绕感兴趣DNA区域的序列进行裂解以用于例如通过DNA测序来进行下游分析。将盒在合适的温度下孵育合适的时间段以允许有效裂解HMW DNA。优选地,由定制的CRISPR-Cas9试剂释放的DNA片段的尺寸小于约5兆碱基,尺寸更优选小于约2兆碱基,从而可以将释放的DNA可靠地与未裂解DNA分离,所述未裂解DNA保持被捕获在样品孔的边缘。
在图3D中,在样品DNA的裂解之后,试剂孔可以被清空并重新填充纯化试剂。在一些实施方案中,纯化试剂与裂解试剂相似或相同。然后可以激活分离电极。因此,随着裂解试剂电泳移动通过样品孔,Cas9-gRNA复合物可随后变性和/或降解。
虽然预计本发明的方法对于长距离基因组分析特别有用,但是该方法可以适用于有靶向选择小(这里的“小”意思是约10至约5000bp的长度)和大(这里的“大”意思是从5000bp到中等单兆碱基对的长度)DNA片段。在任一种情况下,一对定制的CRISPR/Cas9裂解试剂被配置用于将被回收用于分析的每个基因组DNA片段。
对于对应于包含具有细胞壁的细胞(例如细菌,真菌和植物)的样品的一些实施方案,将样品加载到等渗试剂混合物中,所述等渗试剂混合物含有配置为消化细胞壁、从而将原始样品细胞转化为原生质球的酶。在一些这样的实施方案中,样品孔在不向试剂孔添加裂解试剂的情况下被加载,使得细胞壁消化可以持续较长时间,而没有机会在电泳纯化步骤之前将裂解试剂扩散到样品孔中。
给出以下实施例以帮助说明和支持本公开的一些实施方案,并且以下实施例不被认为是限制性的。
实施例1:用于从HLA基因座分离长DNA片段的定制Cas9gRNA裂解酶试剂。
背景.HLA分子由类别I(HLA-A,-B,-C)和II(HLA-DRB1/3/4/5,-DQB1,-DPB1)基因编码。这些基因产生不同的肽给T细胞受体,其调节T细胞发展,自我耐受和适应性免疫。HLA分子是免疫原性的,并且可以成为同种异体移植环境中的细胞和体液免疫应答的靶标,因此HLA分型已成为评估移植供体和受体之间免疫学相容性的护理标准。已经使用Sanger测序进行了单个碱基分辨率的HLA分型,这是劳动力密集且昂贵的。由于大量的单核苷酸变异体(SNV)需要被分为单独的单倍型,因此经常出现顺式-反式模糊性,其只能通过包括具有序列特异性引物的PCR的额外测试来解析。包括Illumina和Ion-torrent在内的几种新一代测序(NGS)技术已经降低了HLA分型的成本,但它们无法直接在超过几百个碱基对的距离上定相SNV。第三代测序仪如Pacific Biosciences(PacBio)和Oxford Nanopore生产的测序仪可以便宜地从单一DNA分子中获得长读数(long read)。原则上,这可以允许HLA基因座的单倍型解析测序;然而,目前还没有建立从基因组中选择特定长DNA片段的方法。这可以通过本申请的发明方法来实现。为此,执行以下步骤。
编码gRNA的寡核苷酸从HLA-A基因座切割长片段。编码gRNA靶向序列和侧翼碱基的DNA寡核苷酸被配置为切割HLA-A基因座的5’和3’末端。一个或多个gRNA可以被靶向至每个末端(这里,我们靶向两个)。在这个实施例中,靶向序列和少量恒定的gRNA序列从IDT技术(IDT technologies)订购,然后被克隆到编码T7启动子下游的gRNA恒定区的DR274载体[PMID:23360964]中。所得质粒经PCR扩增并进行体外转录反应以产生gRNA,产生以下序列:5’ggNNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTT,
其中“N’”代表gRNA靶向序列,并且这些N’的5’的2个g是T7转录起始所需的。
例如,为了靶向HLA-A基因,我们使用以下靶向序列:对于HLA-A的gRNA 5’:
5HLA-A-TS1
5’-GAAAAGAACAGTTACGTAGC-3’,它恰好位于AGG PAM序列的上游并且位于染色体6chr6:29,941,185-29,941,204(所有坐标来自人类参考基因组装配版本GRCh38)。
5HLA-A-TS2
5’-CCAGAAGCTTCACAAGACCG-3’,它恰好位于AGG PAM的上游,位于chr6:29,941,096-29,941,115。靶位点5HLA-A-TS1和5HLA-A-TS2被配置为裂解基因组位点,所述基因组位点分别在5’侧约1350和1440bp侧接共有HLA-A编码起始点(chr6:29,942,553)。
3HLA-A-TS1
5’-ATTCCTTATATTCACCCCCA-3’它恰好位于GGG PAM的上游,并且位于chr6:29949521-29949540
3HLA-A-TS2
5’-CATTCCTTATATTCACCCCC-3’它恰好位于AGG PAM的上游,并且位于chr6:29949520-29949539
3HLA-A-TS1和3HLA-A-TS2彼此重叠并且被配置为在3’侧约4090bp侧接HLA-A共有编码区(chr6:29,945,453)的末端的位置裂解。
预测来自这些HLA-A靶位点的靶向Cas9裂解产物的长度约为~8330bp(5HLA-A-TS1至2HLA-A-TS1或2)和8420bp(5HLA-A-TS2至3HLA-A-TS1或2)。
为了将这些靶向序列中的每一个克隆到DR274载体[PMID:23360964]中,对每个靶向序列订购两个引物并一起退火。这些引物包括来自gRNA恒定部分或侧翼T7启动子的一些序列以促进克隆,并且如下:
5HLA-A-TS1F 5’-TAGG GAAAAGAACAGTTACGTAGC-3
5HLA-A-TS1R 5’-AAAC GCTACGTAACTGTTCTTTTC-3
5HLA-A-TS2F 5’-TAGG TTGAAAGCAGCAGAATTCTT-3
5HLA-A-TS2R 5’-AAAC AAGAATTCTGCTGCTTTCAA-3
3HLA-A-TS1F 5’-TAGG ATTCCTTATATTCACCCCCA-3
3HLA-A-TS1R 5’-AAAC TGGGGGTGAATATAAGGAAT-3
3HLA-A-TS2F 5’-TAGG TCTATCAACAAATTGCTAGG-3
3HLA-A-TS2R 5’-AAAC CCTAGCAATTTGTTGATAGA-3
将gRNA编码寡核苷酸克隆到具有T7启动子的载体中。用BsaI切割质粒载体DR274,将其在琼脂糖凝胶上纯化,并使用Qiagen凝胶纯化试剂盒进行清洁。在以下反应中将100uM每种gRNA编码寡核苷酸退火至其互补物:
该反应物在100℃下加热3-5分钟。之后,加热块被关闭并使其冷却。然后使用以下反应使退火的寡核苷酸磷酸化:
通过温和涡旋将该反应物混合并在37℃孵育30分钟。接下来,使用以下反应将退火的磷酸化寡核苷酸连接到编码T7启动子的质粒中:
在15℃过夜,或在室温2小时。
接下来,将1ul质粒混合物转化到大肠杆菌中,并将其在补充有氨苄青霉素的丰富培养基(LB)琼脂板上铺板。选择Ampr克隆,并通过菌落PCR和Sanger测序验证含有所需指导RNA序列的质粒。将正确的克隆在1ml LB+amp液体培养基中生长,在37℃过夜,并使用Qiagen DNA纯化试剂盒从培养物中分离质粒。
来自质粒模板的gRNA的PCR扩增。为了从质粒模板产生gRNA,将质粒的T7启动子和gRNA区通过PCR进行扩增,然后用作体外转录反应的模板。PCR引物如下:
正向4989GTTGGAACCTCTTACGTGCC
反向5008AAAAGCACCGACTCGGTG。PCR反应设置如下:
该反应循环如下:98℃30s 1个循环(98℃,10s,60℃30s,72℃30s×35个循环),72℃5min,4℃无限地。
该PCR将产生369bp的PCR片段,其然后用Qiagen柱纯化。
使用Mmessage Mmachine T7体外转录试剂盒(AMBION CAT#1344)的RNA合成反应。为了从DNA片段产生gRNA,如下进行体外转录反应:
添加H20到20ul
在37℃孵育4hr
为了回收RNA,加入0.5ul Turbo DNAse(2单位/μl)并在37℃孵育15分钟。然后加入30ul 50mM EDTA pH8.0。加热至80℃持续15分钟以杀死DNAse并使用BIO-RAD Micro Bio-Spin柱(cat#732-6250)回收RNA。通过填充500ul的TE并以1000g旋转2分钟来平衡TE中的Micro Bio-Spin P30柱。然后将50ul样品加载到柱上,以1000g旋转4分钟。将样品洗脱~50ul。gRNA的浓度应为约~200ng/ul,总产量~10ug的RNA。
定制功能性Cas9-gRNA复合物的体外重建。为了形成活性gRNA-Cas9复合物,将2.5ul的cas9蛋白(3.18ug/ul,New England Biolabs cat#M0386M(20uM cas9蛋白))与10ul RNA(2000ng)在总共80ul的1X NEB缓冲液4(New England Biolabs,50mM乙酸钾,20mM Tris-醋酸盐,10mM乙酸镁,1mM DTT,pH7.9)中混合。37℃预孵15分钟。重构的cas9浓度为0.63uM(0.1ug/ul)。
实施例2.使用本发明的电泳方法从人HLA基因座靶向切除和回收长DNA片段。
该实施例使用'833PCT中描述的制备型电泳系统,其实例也显示在图2中,右侧。为了简化说明,图3A-E示出了该实例的示意性工作流程,其中仅示出了分离凝胶和洗脱模块。使用在0.5XKBB缓冲液(51mM Tris(碱),29mM TAPS(酸),0.1mM EDTA(酸),pH8.7)中含有0.75%琼脂糖的本发明盒。试剂和样品孔的总体积分别为350和90ul。
通过选择性裂解RBC从全血制备人WBC。所有步骤均在室温下进行。向12mL全血(ACD抗凝剂)中加入36mL红细胞(RBC)裂解缓冲液(155mM氯化铵;10mM NaHCO3;1mM Na2EDTA)。摇动该溶液3分钟,并通过400xg离心4分钟沉淀白色细胞。倾倒出粉红色的上清液,通过在25mL RBC裂解缓冲液中涡旋使红色沉淀重新悬浮。第二次旋转并倾析后,将粉红色沉淀重新悬浮于900uL RBC裂解缓冲液中。
通过Qubit测量WBC DNA浓度。使用Qubit HS分析(Life Technologies)。通过以下来裂解WBC:将40uL的WBC与160uL的TE/50mM NaCl/1%SDS混合,然后在65℃孵育3分钟。加入TE(800uL),涡旋以降低粘度后,按照供应商的方案,将1或2.5uL DNA加入到199uL Qubit试剂中。
通过定制-gRNA-Cas9试剂和制备型电泳靶向回收HLA片段。将纯化的WBC(含有10-12ug基因组DNA)加载到RBC裂解缓冲液中的盒的空样品孔中。装载的样品的总体积为80ul。将320ul的裂解缓冲液(10mM Tris-HCl,pH7.5,1mM EDTA,3%SDS,5%甘油,各50ug/mL溴酚蓝和酚红)加入到空的试剂孔中。参见图3A。
通过在SageELF仪器(Sage Science,Inc.)中使用100V的分离电极电泳40分钟进行细胞裂解和DNA纯化。参见图3B。
纯化电泳后,试剂和样品孔被清空。将试剂孔重新加载320ul 1X NEB缓冲液4。将样品孔重新加载80μl的1XNEB缓冲液#4,其含有实施例1中所述的重构的定制Cas9-gRNA试剂混合物,所述混合物浓度为总体积80ul中的5pM(Cas9蛋白质的总浓度)。盒在37℃孵育60分钟以允许固定的基因组DNA内的HLA靶位点的裂解。见图3C。
裂解后,将试剂孔和样品孔清空,用320μl 0.5XKBB+3%十二烷基肌氨酸钠重新填充试剂孔。样品孔中装有80μl相同的缓冲液,其另外含有200ug/ml蛋白酶K。该盒在37℃孵育30分钟以允许消化Cas9蛋白试剂(参见图3D)。
消化后,盒在SageELF仪器中以分离模式使用60V连续场的程序电泳1小时。参见图3E。
分离电泳后,使用50V的电压在ELF仪器中进行电洗脱45分钟。在洗脱结束时,反向施加25V电场5秒钟,以帮助从洗脱模块的超滤膜释放洗脱的DNA。可通过手动或自动液体处理装置从电泳缓冲液中的洗脱模块中去除目标片段(洗脱和从洗脱模块中回收未在图3A-E中示出)。
实施例3:用于靶向分离人蛋白质编码序列的定制gRNA-Cas9构建体(外显子分离)。
指导RNA设计。我们设计/构建了用于将所有人类外显子(连同外显子每一侧上的一些侧翼非外显子序列)裂解成长度为200-500bp的片段的gRNA。通过检查紧接NGG位点5’的所有20bp DNA序列挑选gRNA,然后从该组中选择处于外显子侧翼的对。对于长于500个碱基对的外显子,gRNA也配置在外显子的内部,以便将外显子序列切成2个或更多个长度小于500个碱基对的片段。丢弃具有准确匹配(exact match)、或在脱靶基因组位点的1或2bp错配的gRNA。
大规模平行寡核苷酸合成和扩增。从Custom Array(Bothell,WA,USA)订购编码gRNA的核苷酸的文库。格式是5’CGCTCGCACCGCTAGCTAATACGACTCACTATAGGNNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTT,
其中“N’”代表靶向序列,加下划线的序列是T7启动子。通过用以下引物进行PCR扩增该文库:正向:5’CGCTCGCACCGCTAGCTAATACGACT-3,和反向5’AAAAAGCACCGACTCGGTGCCACTTTT-3’。预期PCR产物是134bp并被凝胶纯化。
使用Mmessage Mmachine T7(AMBION CAT#1344)通过体外转录产生gRNA。为了从DNA片段产生gRNA,如下进行体外翻译反应:
添加H20到20ul
在37℃孵育4hr
为了回收RNA,加入0.5μl Turbo DNAse(2单位/μl)并在37℃孵育15分钟。然后加入30μl 50mM EDTA pH 8.0。加热至80℃持续15分钟以杀死DNAse,并使用BIO-RAD Micro-Bio-spin柱(CAT#732-6250)回收RNA。通过填充500ul TE并以1000g旋转2分钟来平衡TE中的Micro Bio-Spin P30柱。然后将50ul样品加载到柱上,以1000g旋转4分钟。将样品洗脱~50ul。gRNA文库的浓度应为~200ng/ul,总产量约为~10ug的RNA。
定制功能性Cas9-gRNA复合物的体外重建。为了裂解基因组DNA,在总共80ul的1X NEB缓冲液4中将2.5ul cas9蛋白(3.18ug/ul,New England Biolabs cat#M0386M(20uM cas9蛋白))与10ul RNA(2000ng)混合(New England Biolabs,50mM乙酸钾,20mM Tris-醋酸盐,10mM乙酸镁,1mM DTT,pH7.9)。37℃预孵15分钟。重构的cas9浓度为0.63uM(0.1ug/ul)。
通过制备型电泳靶向切除和纯化含外显子的gDNA。如实施例2中所述纯化来自人WBC的总基因组DNA。如实施例2中所述进行靶向切除,除了两个例外:
(1)裂解步骤使用更高浓度的重构的gRNA-Cas9复合物:浓度为0.63μM的80ul重构的gRNA-Cas9复合物,这反映了切除所有含外显子的DNA所需的裂解的~20,000倍更高数量的裂解,其相对于实施例2中用于HLA-A情况的8个位点。
(2)制备型凝胶盒使用2%琼脂糖凝胶(代替实施例2中使用的0.75%凝胶),为了预期的200-500bp靶的更好的尺寸分辨。
实施例4-通过靶向基因组DNA测序表征易位断裂点
细胞遗传学研究导致发现特定的染色体重排与人类肿瘤相关。特定人类癌症中第一个可再现的染色体异常是与慢性粒细胞白血病有关的费城染色体。后来的分子研究表明,这种重排导致产生融合蛋白,其由c-abl,v-abl致癌基因的同系物和称为bcr(断裂点簇区域)的基因组成。这种bcr-abl融合蛋白是致癌的并且是非常成功的称为格列卫的治疗剂的靶标。
现在有数百个(如果不是数千个)与各种肿瘤相关的染色体易位。染色体断裂的常见部位发生在编码间变性淋巴瘤激酶(ALK)编码基因中,位于染色体2p23。它在许多不同的肿瘤中易位到多种不同的染色体,PMID:23814043。然而,它在非霍奇金淋巴瘤的一个子集中最常被重新排列,其中在染色体5q35上涉及ALK和核磷蛋白(NPM1)基因的易位被观察到,PMID:25869285。该易位允许形成NPM1-ALK融合基因,该基因是治疗剂克唑替尼(crizotinib)的靶[PMID:24491302]。这些易位中断裂的确切位置是可变的,因此很难被传统的NGS检测到。获得含有重排基因座的大DNA片段以用于序列分析的能力将减少对费力的细胞遗传学测定的需要。
编码gRNA的寡核苷酸切割包含NPM1-ALK易位的长片段。为了分离该片段,编码gRNA的DNA寡核苷酸被配置成切除染色体5q35上的NPM1基因的5’和染色体2p23上的ALK基因的3’。在具有NPM1-ALK t(2;5)易位的细胞中,这些gRNA将指导Cas9以切割含有重排连接的大片段。可以将两个或更多个gRNA靶向每个末端以确保在指定区域的切割和含有NPM1-ALK的片段的切除。
编码靶向序列的短寡核苷酸被克隆到编码T7启动子下游的gRNA恒定区的DR274载体[PMID:23360964]中。在体外转录后,产生具有以下序列的gRNA:体外转录后,产生具有以下序列的gRNA:
5’ggNNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAAAT AGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTT,
其中“N’”代表gRNA靶向序列,并且5’g是T7转录所需的。
在这个例子中,我们使用以下靶向序列:
对于NPM1的5’侧的gRNA:
NPM1-TS1
5’CAAGTCACCCGCTTTCTTTC 3’,它恰好位于AGG PAM序列的上游并且位于chr5:171,387,031-171,387,050,其在NPM1编码序列(chr5:171,387,949)的起始处上游约900bp处,并且在910bp长NPM1内含子5(chr5:171,391,799)的起始处上游约4750bp处,其中发生许多NPM1-ALK断裂点。
NPM1-TS2
5’GACTTTGGAGATGTTTTCTC3’它恰好位于AGG PAM的上游,并且位于chr5:171,387,185-171,387,204,其在NPM1编码序列起始处上游约750bp处,以及NPM1内含子5起始处上游~4600bp处。
对于ALK 3’侧的gRNA:
ALK-TS1
5’-GAAGAAAACATGGCACAAAT-3’它恰好位于GGG PAM的上游并且位于chr2:29,190,272-29,190,291,其距离ALK编码序列(chr2:29,193,225)末端下游(3’)2930bp处,和距离1940-bp长ALK内含子19的末端下游(3’)33240bp处,其中发生许多NPM1-ALK断裂点。
ALK-TS2
5’-CAATGGGTCAGATAACTCAA-3’它恰好位于GGG PAM的正上游并且位于chr2:29,190,518-29,190,537,其距离ALK编码序列末端下游(3’)~2700bp。
图4显示了靶向位点,相关编码区和产生NPM1-ALK融合重排的一些共同断裂点的示意图(Morris等,(1994)Science 263:1281;Duyster,等,(2001)Oncogene 20:5623)。
考虑到由于融合的任一末端上的备选gRNA靶位点以及910bp NPM1内含子5和1940bp ALK内含子19内的易位断裂点位置的变化引起的片段尺寸的可能变化,采用上述定制gRNA-Cas9复合物的NMP1-ALK融合基因的消化应产生长度在37.5kb和40.8kb之间的片段。
为了将这些靶向序列中的每一个克隆到DR274载体中,针对每个靶向序列订购两个引物并一起退火。这些引物包括来自gRNA恒定部分或侧翼T7启动子的一些序列以促进克隆,并且如下:
NPM1-TS1F 5’-TAGGCAAGTCACCCGCTTTCTTTC-3
NPM1-TS1R 5’-AAACGAAAGAAAGCGGGTGACTTG-3
NPM1-TS2F 5’-TAGGACTTTGGAGATGTTTTCTC-3
NPM1-TS2R 5’-AAACGAGAAAACATCTCCAAAGT-3
ALK-TS1F 5’-TAGGAGGGGCGCCCAATTTTGTCT-3
ALK-TS1R 5’-AAACAGACAAAATTGGGCGCCCCT-3
ALK-TS2F 5’-TAGGTCTATCAACAAATTGCTAGGAGG-3
ALK-TS2R 5’-AAAC CCTAGCAATTTGTTGATAGA-3
将编码gRNA的寡核苷酸克隆到具有T7启动子的载体中。用BsaI切割质粒载体DR274[ref],在琼脂糖凝胶上纯化,并用Qiagen凝胶纯化试剂盒清洁。在以下反应中将100uM每种gRNA编码寡核苷酸退火至其互补物:
该反应物在100℃下加热3-5分钟。之后,将加热块关闭并使其冷却。然后使用以下反应使退火的寡核苷酸磷酸化:
通过温和涡旋将该反应物混合并在37℃孵育30分钟。
接下来,使用以下反应将退火的磷酸化寡核苷酸连接到编码T7启动子的质粒中:
在15℃过夜,或在室温2小时。
接下来,将1μl质粒混合物转化到大肠杆菌中,并在补充有氨苄青霉素的丰富培养基(LB)琼脂板上进行铺板。选择Ampr克隆,并通过菌落PCR和Sanger测序验证含有所需指导RNA序列的质粒。将正确的克隆在1ml LB+amp液体培养基中生长,在37℃过夜,并使用Qiagen DNA纯化试剂盒从培养物中分离质粒。
来自质粒模板的gRNA的PCR扩增。为了从质粒模板产生gRNA,通过PCR扩增质粒的T7启动子和gRNA区,然后用作体外转录反应中的模板。PCR引物如下:正向4989GTTGGAACCTCTTACGTGCC,反向5008AAAAGCACCGACTCGGTG。PCR反应设置如下:
该反应循环如下:98℃30s 1个循环(98℃,10s,60℃30s,72℃30s×35个循环),72℃5min,4℃无限地。该PCR将产生369bp的PCR片段,其然后用Qiagen柱纯化。
使用Mmessage Mmachine T7体外转录试剂盒(AMBION CAT#1344)的RNA合成反应。为了从DNA片段产生gRNA,如下进行体外转录反应:
37℃孵育4小时
为了回收RNA,加入0.5ul的Turbo DNAse(2单位/ul)并在37℃孵育15分钟。然后加入30ul 50mM EDTA pH8.0。加热至80℃持续15分钟以杀死DNAse并使用BIO-RAD Micro Bio-Spin柱(caT#732-6250)回收RNA。通过填充500ul TE并以1000g旋转2分钟来平衡TE中的Micro Bio-Spin P30柱。然后将50ul样品加载到柱上,以1000g旋转4分钟。样品将洗脱~50ul。gRNA的浓度应为~200ng/ul,用于总产量~10ug的RNA。
定制功能性Cas9-gRNA复合物的体外重建。为了形成活性gRNA-Cas9复合物,将2.5ul的cas9蛋白(3.18ug/ul,New England Biolabs cat#M0386M(20uM cas9蛋白))与10ul RNA(2000ng)在总共80μl的1X NEB缓冲液4(New England Biolabs,50mM乙酸钾,20mM Tris-醋酸盐,10mM乙酸镁,1mM DTT,pH7.9)中混合。37℃预孵15分钟。重构的cas9浓度为0.63uM(0.1ug/ul)。
通过制备型电泳靶向切除和纯化基因组NPM1-ALK融合片段。如实施例2中所述纯化来自人WBC's的总基因组DNA。使用NPM1-ALK易位特异性Cas9靶向复合物的靶向切除也如实施例2所述进行。
消化后,将盒在SageELF仪器中以分离模式使用60V连续场的程序进行1小时电泳,然后使用波形进行2小时的时间段脉冲场电泳(在80V下)以解析5-430kb DNA,如Pippin Pulse用户手册(http://www.sagescience.com/product-support/pippin-pulse-support/)中所述。
分离电泳后,使用50V的电压在ELF仪器中进行电洗脱45分钟。在洗脱结束时,反向施加25V电场5秒钟,以帮助从洗脱模块的超滤膜释放洗脱的DNA。可通过手动或自动液体处理装置从电泳缓冲液中的洗脱模块中去除目标片段(洗脱和从洗脱模块中回收未示出在图3A-E中)。
在本申请的任何地方呈现的对出版物或其他文件(包括但不限于专利,专利申请,文章,网页,书籍等)的任何和所有引用均通过引用将所述出版物或其他文件整体并入本文。
如其他地方所述,所公开的实施方案仅出于说明的目的而呈现,而不是限制性的。其他实施方案是可能的并且由本公开内容覆盖,根据本文包含的教导,这将变得显而易见。因此,本公开的宽度和范围不应受任何上述实施方案的限制,而应仅根据本公开所支持的权利要求及其等同物来限定。此外,本发明的实施方案可以包括,可以进一步包括来自任何其他公开的方法、组合物、系统和装置的任何和全部元素的方法,组合物、系统和装置/设备,所述元素包括对应于从生物样品中分离核酸的任何和全部元素(例如,包含核酸和非核酸元素)。换句话说,来自一个或另一个公开的实施方案的元素可以与来自其他公开的实施方案的元素互换。此外,可以通过以下来实现一些其他实施方案:将本文公开的一个和/或另一个特征与通过引用并入的材料中公开的方法、组合物、系统和设备及其一个或多个特征组合。另外,所公开的实施方案的一个或多个特征/元素可以被移除并且仍然导致可授予专利的主题(并且因此导致本主题公开的更多实施方案)。此外,与现有技术的教导相比,一些实施方案对应于特别缺少一个和/或另一个元素、结构和/或步骤(如适用)的方法、组合物、系统和设备,并且因此代表可授予专利的主题并且可以从中区分(即,针对这些实施方案的权利要求可能包含负面限制以指出缺少现有技术教导的一个或多个特征)。
当描述核酸加工时,诸如链接、结合、连接、附接、相互作用等的术语应该被理解为指的是引起被引用的元素的联结的连接,无论这种联结是永久的或可能是可逆的。这些术语不应被理解为需要形成共价键,尽管可能形成共价类型键。
如本文所定义和使用的所有定义应理解为以以下为准:字典定义,通过引用并入的文件中的定义和/或所定义的术语的普通含义。
如本文中在说明书和权利要求书中使用的不定冠词“一”和“一个”,除非有明确的相反指示,应理解为表示“至少一个”。
如本文在说明书和权利要求书中使用的短语“和/或”应理解为意指如此结合的元素的“一个或两个”,即在一些情况下结合地存在的和在其他情况下分离地存在的元素。用“和/或”列出的多个元素应该以相同的方式解释,即如此结合的元素中的“一个或多个”。除了由“和/或”子句明确标识的元素之外的其他元素可以可选地存在,不管所述其他元素与具体标识的那些元素相关还是不相关。因此,作为非限制性示例,当与诸如“包括”的开放式语言结合使用时,对“A和/或B”的引用在一个实施例中可以仅指A(可选地包括不是B的元素);在另一个实施方案中,仅指B(可选地包括除A之外的元素);在又一个实施方案中,指A和B(可选地包括其他元素);等等
如在本说明书和权利要求书中所使用的,“或”应当被理解为具有与以上定义的“和/或”相同的含义。例如,当分离列表中的项目时,“或”或“和/或”应被解释为包含性的,即包含至少一个,但也包括多个元素或元素的列表中的多于一个,以及可选的其他未列出项目。只有明确相反指出的术语,例如“仅一个”或“恰好一个”,或者当在权利要求中使用时,“由...组成”将指包含多个元素或元素列表中的恰好一个元素。一般而言,在此使用的术语“或”当在排他性方面在“或”之前有诸如“任一”,“中的一个”,“中的仅一个”或“中的正好一个”时,应当仅被解释为指示排他性替代(即“一个或另一个,但不是两个”)。当用于权利要求中时,“基本上由...组成”应具有其在专利法领域中使用的普通含义。
如本文中在说明书和权利要求书中所使用的,关于一个或多个元素的列表的短语“至少一个”应该理解为是指选自元素列表中的任何一个或多个元素中的至少一个元素,但不一定包括元素列表内具体列出的每个和各个元素中的至少一个,并且不排除元素列表中的元素的任何组合。该定义还允许,可以可选地存在除在短语“至少一个”所指的元素列表内具体标识的元素之外的元素,不管该元素与具体标识的那些元素相关还是不相关。因此,作为非限制性示例,“A和B中的至少一个”(或者等同地,“A或B中的至少一个”,或者等同地“A和/或B中的至少一个”)可以在一个实施方案中,意指至少一个,可选地包括多于一个A,而不存在B(并且可选地包括除了B之外的元素);在另一个实施方案中,意指至少一个,可选地包括多于一个B,而不存在A(并且可选地包括除了A之外的元素);在又一个实施方案中,意指至少一个A,并且至少任选地包括多于一个B(并且可选地包括其他元素);等等。
在权利要求以及以上说明书中,诸如“包含”,“包括”,“携带”,“具有”,“含有”,“涉及”,“保留”,“组成”等的所有过渡短语应理解为是开放式的,即意味着包括但不限于。只有过渡短语“由......组成”和“基本上由...组成”应分别是封闭式或半封闭式过渡短语,如美国专利局专利审查程序手册第2111.03节所述。
- 还没有人留言评论。精彩留言会获得点赞!