一种法匹拉韦的合成方法与流程
本发明属于药物合成领域,具体涉及法匹拉韦的新制备方法。
背景技术:
法匹拉韦(favipiravir),化学名为6-氟-3-羟基吡嗪-2-甲酰胺,分子式为:C5H4N3O2F,分子量为:157.1,具有如下结构式:
法匹拉韦由日本富山化学有限公司研究开发,2011年,在日本完成III期临床试验,2014年批准上市,临床上主要用于流感治疗,它是RNA依赖的RNA聚合酶抑制剂类的广谱抗病毒药物。研究表明,法匹拉韦在细胞内酶的作用下形成法匹拉韦-呋喃核糖基-5-三磷酸(T-705RTP),竞争性地抑制病毒RNA依赖的RNA聚合酶,从而抑制病毒基因组复制和转录;同时也可以浸入到病毒基因,诱发突变发挥抗病毒作用。
目前,国内外法匹拉韦的主要合成方法大致如下:
路线一:专利WO00/01569报道,用6-溴-3-氨基吡嗪-2-甲酸甲酯经重氮化后用甲醇醇解、钯催化下氨基取代及氨解制得6-氨基-3-甲氧基吡嗪-2-甲酰胺,经重氮化及氟取代,然后在三甲基硅烷和碘化钠作用下得到目标化合物(合成线路见方程式1)。此法将氨基取代反应中所使用的催化剂(二亚苄基丙酮)二钯和(S)-(-)-2,2′-双(二苯膦基)-1,1′-联萘价格较昂贵,且在最后一步反应中用三甲基氯硅烷和碘化钠去甲基化反应难以控制,收率低,不适合于工业生产。另外,该合成线路的最后需要制备成二环己胺盐,成盐之后再进行腈水解反应,两步的收率为26.4%,且水分对二环己胺成盐反应的影响很大,水分过多则不易成盐,导致收率降低,对实验操作要求较高。
方程式1
路线二:专利CN102775358A报道,用3-氨基-2-吡嗪甲酸作为起始原料经过羟基化、酯化、胺化、硝化、还原、氟化六步反应制得目标化合物(合成线路见方程式2).虽然反应路线较短,但由于6-硝基-3-羟基-2-吡嗪酰胺在一般有机溶剂中溶解度有限,将其还原成氨基化合物较困难,该步骤使用了昂贵钯试剂,总收率较低。
方程式2
路线三:李行舟等以氨基丙二酸二乙酯盐酸盐为原料,经中和、氨解、环合、溴化、氯代及脱水、氟化、选择性水解、成盐纯化、水解得到法匹拉韦。该路线较长,收率较低,只有9%(合成线路见方程式3)。肖新荣等同样以氨基丙二酸二乙酯盐酸盐为起始原料,氨水氨解后与乙二醛环合直接制得3-羟基-2-吡嗪酰胺、经硝化、氯代、氟代、水解成盐后、再经过氧化氢氧化,制得法匹拉韦。该法收率偏低,只有5.6%.专利US 8835636有类似报道,只不过是以氨基丙二酰胺为原料,经环合、溴化、氯代及脱水、氟化、3位-选择性水解、腈水解得到法匹拉韦。
路线四:Fangyuan Shi等以3-羟基吡嗪-2-羧酸为原料,经过酯化、氨解、硝化、还原、氟化6步合成法匹拉韦(合成线路见方程式4),此路线中有硝化反应,对设备要求较高,收率较低。
路线五:CN104496917以6-溴-3-氨基吡嗪-2-羧酸为原料,经过酯化、在重氮化水解反应、羟基保护、氟代、脱保护、氨化得到法匹拉韦(合成线路见方程式5)。此路线使用Pd催化剂脱保护,成本较高。
综上分析,现有合成方法中还存在诸多技术问题,难于适用于工业化生产。
技术实现要素:
为克服现有合成方法工艺复杂、原料昂贵、收率低等技术问题,本发明提供了一种法匹拉韦的合成方法,旨在简化操作工艺,提升目的产物产率,达到适用于工业生产的目的。
在工业上,对于药物的合成而言,主要关注点除收率外,还需要重点关注目的产物的杂质含量;特别是某些关键中间体、关键杂质的含量。为解决关键杂质问题,现有技术常采用色谱纯化方式,该类处理方法存在溶剂用量大、效率低、收率低等一系列缺陷,难于适合工业化生产需求;需求一种无需色谱纯化、且产物收率高,目的产物质量可控的合成方法就显得尤为重要;本发明人通过大量研究,开发出一种无需色谱纯化、产品质量可控,收率高的法匹拉韦的合成方法,如下所示:
一种法匹拉韦的合成方法,包括以下步骤:
步骤(1):3-氨基吡嗪-2-羧酸(1)与醇发生酯化反应,得到3-氨基吡嗪-2-羧酸酯(2);
步骤(2):将步骤(1)制得的3-氨基吡嗪-2-羧酸酯与NBS进行溴化反应,溴化反应得到的粗产品经二氯甲烷打浆后经醇重结晶,制得6-溴-3-氨基吡嗪-2-羧酸酯(3);
步骤(3):将步骤(2)制得的6-溴-3-氨基吡嗪-2-羧酸酯与亚硝酸盐在酸性条件下进行重氮化反应,后处理得到6-溴-3-羟基吡嗪-2-羧酸酯(4);
步骤(4):将步骤(3)制得的6-溴-3-羟基吡嗪-2-羧酸酯在氨水中进行氨解反应,经后处理得到6-溴-3-羟基吡嗪-2-甲酰胺(5);
步骤(5):将步骤(4)制得的6-溴-3-羟基吡嗪-2-甲酰胺与三氯氧磷、在缚酸剂存在下进行氯代-脱水反应,氯代-脱水反应的粗品经重结晶处理制得3,6-二氯吡嗪-2-甲腈(6);
步骤(6):将步骤(5)制得的3,6-二氯吡嗪-2-甲腈与氟化剂进行芳环氟代反应;将氟代反应产物直接经双氧水催化,进行氰基水解反应;氰基水解反应产物直接经碱水溶液催化,进行芳环羟基取代反应,随后再经纯化处理,制得法匹拉韦(7)。
本发明制备方法,在合成线路方面,本发明独创性地采用步骤(6)顺次进行的芳环氟代反应、双氧水氰基水解反应和芳环羟基取代反应的串联合成思路,可明显简化制备流程,还可明显控制目的产物的杂质含量和收率。此外,再协同配合采用本发明所述的重结晶的方法,对合成线路的关键中间体3和6进行纯化,相较于现有的色谱纯化方式,本发明方法操作更简便,且可控制中间体3和6的关键杂质,产物的收率更高;尤其适合于工业放大生产。
本发明的合成线路见方程式6
步骤(1)中,将3-氨基吡嗪-2-羧酸(1)与醇在酸催化下进行酯化反应。
作为优选,所述的醇为甲醇、乙醇、丙醇中的一种;进一步优选为甲醇。
步骤(1)中,所述的酸优选为浓硫酸。
作为优选,步骤(1)中,将3-氨基吡嗪-2-羧酸(1)和醇在浓硫酸催化下进行酯化反应。
作为优选,步骤(1)中,3-氨基吡嗪-2-羧酸、醇的重量比为1∶6~9。
进一步优选,步骤(1)中,3-氨基吡嗪-2-羧酸、甲醇的重量比为1∶6~9。
作为优选,步骤(1)中,3-氨基吡嗪-2-羧酸、浓硫酸的重量比比为1∶1~∶3;
所述摩尔量的浓硫酸优选缓慢投加,作为优选,浓硫酸的滴加的时间为0.5~3h。
作为优选,步骤(1)中,酯化反应优选在室温下进行,例如20~35℃。
作为优选,步骤(1)中,酯化反应时间为36~60h。
酯化反应结束后,优选采用弱碱性化合物优选为碳酸氢钠调节体系的pH为7-8。随后采用乙酸乙酯萃取,无水硫酸钠干燥,经减压蒸馏除去乙酸乙酯,50℃真空干燥2h得到棕色固体。
步骤(2)中,将步骤(1)制得的3-氨基吡嗪-2-羧酸酯与NBS优选在乙腈的溶剂氛围下进行溴化反应。
作为优选,3-氨基吡嗪-2-羧酸酯与NBS的投加摩尔比为1∶1.0~1.1。
所述投加量的NBS优选分批投加,且分批投加的间隔时间优选为2h。
3-氨基吡嗪-2-羧酸酯与乙腈的投加重量比为1∶7~10。
作为优选,步骤(2)优选在室温下进行,例如优选为20~35℃。
作为优选,步骤(2)中,溴化反应时间为20~30h。
溴化反应结束后,采用碱液调整体系的pH,例如采用碳酸氢钠调整反应液体系的pH为7-8。随后用乙酸乙酯萃取,无水硫酸钠干燥,经减压蒸馏除去乙酸乙酯,得到6-溴-3-氨基吡嗪-2-羧酸酯粗品。
本发明人发现,6-溴-3-氨基吡嗪-2-羧酸酯3为本线路的关键中间体。本发明人发现在NBS催化下的溴代反应不易反应完全,从而使得生成的6-溴-3-氨基吡嗪-2-羧酸酯粗品中含有部分未反应的中间体2,发生溴代反应的同时有可能生成与3极性接近的5-溴-3-氨基吡嗪-2-羧酸甲酯,或者产生5,6-二溴-3-氨基吡嗪-2-羧酸甲酯。如不提纯,其中的杂质将会直接引入目标产物中,进而影响目标产物的质量。鉴于该中间体和上述杂质的特性,现有制备方法常采用柱色谱的方法对其进行纯化,然而,采用色谱纯化的制备效率很低、成本很高,且产物的损耗比较大,现有的方法存在较多技术难题,严重阻碍了工业生产。
本发明可通过调节加入NBS的含量及反应时间,控制这些杂质的含量;同时,本发明人尝试采用结晶的方法对中间体3进行纯化处理,例如,本发明人采用DCM(二氯甲烷)、EA(乙酸乙酯)、甲醇、乙醇等溶剂进行结晶处理,结果发现,都存在提纯困难等技术问题;通过深入研究发现,本发明人终于发现了采用二氯甲烷-醇体系可达到良好的结晶效果。通过本步的重结晶可以将中间体2及其它杂质除去,进而保证了目的产物的质量。
作为优选,步骤(2)中,预先将6-溴-3-氨基吡嗪-2-羧酸酯粗品(3的粗品)用DCM打浆,进行一次除杂处理,随后固液分离,除杂后的溶液浓缩后再采用醇进行重结晶,制得3的纯品。
作为优选,步骤(2)中,重结晶过程采用的醇为甲醇、乙醇、异丙醇、叔丁醇中的至少一种;优选为乙醇。
作为优选,6-溴-3-氨基吡嗪-2-羧酸酯粗品与DCM的重量比为1∶25~50。
除杂后的溶液浓缩得到的浓缩物与醇的重量比为1∶5~30。
步骤(2)中,重结晶过程中,析晶温度为25~30℃。
步骤(3)中,亚硝酸盐优选为亚硝酸根的碱金属盐;例如为亚硝酸钠。
步骤(3)中,重氮化反应优选在浓硫酸的催化下进行。
作为优选,步骤(3)中,6-溴-3-氨基吡嗪-2-羧酸酯(3)与亚硝酸盐的摩尔比为1∶1.5~2.5。
作为优选,6-溴-3-氨基吡嗪-2-羧酸酯(3)与浓硫酸的摩尔比为1∶15~20。
作为优选,重氮化反应过程中,预先将6-溴-3-氨基吡嗪-2-羧酸酯(3)与浓硫酸混合,随后在低温下,优选在-5~0℃下分批加入亚硝酸盐;亚硝酸盐投加完成后,升温至室温下搅拌反应。亚硝酸盐投加完成后,优选在20~35℃下搅拌反应。
作为优选,重氮化反应的时间为2.0~4h。
重氮化反应结束后,向反应体系中投加水,优选为冰水或冰块,继续搅拌反应1~2h;
加水反应后,用EA萃取,无水硫酸钠干燥,经减压蒸馏除去乙酸乙酯,50℃真空干燥2h得到浅黄色固体(6-溴-3-羟基吡嗪-2-羧酸酯;4)。
步骤(4)中,将步骤(3)制得的6-溴-3-羟基吡嗪-2-羧酸酯在氨水中进行氨解反应,作为优选,6-溴-3-羟基吡嗪-2-羧酸酯与氨水的重量比为1∶8~15。
步骤(4)中,氨解反应优选在室温下进行,例如为20~35℃。
作为优选,步骤(4)中,氨解反应的时间为2~5h。
氨解反应结束后,向反应体系中投加水,随后采用EA进行萃取,萃取得到的有机相再用无水硫酸钠干燥、随后再浓缩得到6-溴-3-羟基吡嗪-2-甲酰胺(5);
步骤(5):将步骤(4)制得的6-溴-3-羟基吡嗪-2-甲酰胺与三氯氧磷、在缚酸剂存在下进行氯代-脱水反应。
作为优选,所述的缚酸剂为三乙胺、二甲基丙胺、N,N-二异丙基乙胺(DIEA)中的至少一种,进一步优选为N,N-二异丙基乙胺。
作为优选,步骤(5)中,6-溴-3-羟基吡嗪-2-甲酰胺与三氯氧磷的摩尔比为1∶3~5。
作为优选,步骤(5)中,6-溴-3-羟基吡嗪-2-甲酰胺与缚酸剂的摩尔比为1∶2~4。
作为优选,步骤(5)反应溶剂体系为氯苯、乙酸乙酯、乙酸正丁酯或者无溶剂,优化为不使用溶剂。
本发明人发现,对步骤(5)的反应温度、及反应时间进行精准调控,有助于改善制得的中间体6的品质。
作为优选,步骤(5)中,向6-溴-3-羟基吡嗪-2-甲酰胺中或者6-溴-3-羟基吡嗪-2-甲酰胺的反应溶剂混合液中投加三氯氧磷(POCl3),并在65~75℃下反应5~30min;随后降温至室温,并投加缚酸剂,再后依次在55~65℃下反应0.5~1.5h、在75~85℃下反应0.5~1.5h、和在95~105℃下反应3.5~4.5h;反应结束后向体系中投加水,固液分离得3,6-二氯吡嗪-2-甲腈粗品。
本发明采用所述的梯度控温反应方式,可为后续的产品精制减轻负担,且可进一步提升制得的3,6-二氯吡嗪-2-甲腈质量,降低目的产物的杂质含量。
进一步优选,步骤(5)中,向6-溴-3-羟基吡嗪-2-甲酰胺中或者6-溴-3-羟基吡嗪-2-甲酰胺的反应溶剂混合液中投加三氯氧磷(POCl3),并在70℃下反应15min;随后降温至室温,并投加缚酸剂,再后依次在60℃下反应1h、在80℃下反应1h、和在100℃下反应4h;反应结束后向体系中投加水,固液分离得3,6-二氯吡嗪-2-甲腈粗品。
中间体6为本线路的重要中间体,其质量将直接影响到终产物的质量。现有技术常采用色谱分离方式对产品进行纯化,然而该纯化方式存在诸多缺陷,例如操作困难,需要大量洗脱溶剂等等,不利于工业生产。
生成关键中间体6过程中,由中间体4到6过程中原料反应可能反应不完全,且在生成6时,可能氯代不完全生成单氯代产物,与目的产物结构相似,极性相近,故而需严格控制这些杂质的含量,通过重结晶可以除去杂质,从而提高了目的产物的质量。本发明人通过大量研究,开发出一种重结晶的办法。
作为优选,步骤(5)中,重结晶所用溶剂为甲醇、二氯甲烷、石油醚中的至少一种。
作为优选,步骤(5)中,重结晶过程的结晶溶剂为石油醚。
进一步优选,步骤(5)中,重结晶过程中,3,6-二氯吡嗪-2-甲腈粗品与重结晶溶剂的重量比为1∶10~30,析晶温度为25~30℃。
进一步优选,步骤(5)中,向6-溴-3-羟基吡嗪-2-甲酰胺中缓慢加入三氯氧磷(POCl3),并在65~75℃下反应5~30min;随后降温至室温,并投加N,N-二异丙基乙胺,其中6-溴-3-羟基吡嗪-2-甲酰胺、三氯氧磷、二异丙基乙胺的摩尔为:1∶3~5∶2~4,随后依次在55~65℃下反应0.5~1.5h、在75~85℃下反应0.5~1.5h、和在95~105℃下反应3.5~4.5h;反应结束后向体系中投加冰水,固液分离得3,6-二氯吡嗪-2-甲腈粗品,后经石油醚重结晶,重结晶过程中3,6-二氯吡嗪-2-甲腈粗品与石油醚的重量比为1∶10~30,析晶温度为25~30℃。
步骤(6)采用串联一锅合成方式制备;所述的串联一锅合成为依次进行所述的反应,且对各步反应的反应体系不进行纯化、分离处理。
步骤(6)中,将步骤(5)制得的3,6-二氯吡嗪-2-甲腈与氟化剂进行芳环氟代反应;无需对氟代反应产物(反应液)进行纯化,直接经双氧水催化,进行氰基水解反应;氰基水解反应产物(反应液)无需进行纯化,直接经碱水溶液催化,进行芳环羟基取代反应,随后经纯化处理,制得法匹拉韦(7)。
本发明人对串联一锅反应的方法的物料及制备的先后循序进行研究发现,采用本发明的芳环氟代反应-氰基水解反应-芳环羟基取代反应可明显降低目的产物的杂质含量;另外,相比于先氟化-羟基化-水解的现有方法,本发明的腈水解可控,且不易水解成羧酸,目标产物分离简单;研究发现,本发明步骤(6)的收率可高达60%以上,比现有的制备方法提升50%以上。
作为优选,步骤(6)中,反应溶剂为二甲基亚砜、1,4-二氧六环、N,N-二甲基甲酰胺、吡啶中的至少一种,进一步优选为二甲基亚砜。
作为优选,步骤(6)中,向中间体6的溶液体系中投加氟化剂和相转移催化剂,进行所述的芳环氟代反应。
所述的氟化剂为F-的盐。
作为优选,所述的氟化剂为氟化钾、氟化铵、四丁基氟化铵、氟化铯等中的至少一种,进一步优化为氟化钾。
所述的相转移催化剂可为本领域所能预料到的常用相转移物料。
作为优选,所述的相转移催化剂为四丁基三溴化铵(TBAB)。
步骤(6)中,在进行所述的芳环氟代反应前,对反应体系,例如溶剂进行除水处理,反应装置采用干燥的保护性气氛进行置换。
例如,步骤(6)所采用的反应溶剂预先采用甲苯除水。
作为优选,3,6-二氯吡嗪-2-甲腈与氟化剂的摩尔比为1∶5~7。
作为优选,3,6-二氯吡嗪-2-甲腈与相转移催化剂的摩尔比为1∶0.05~0.5。
作为优选,步骤(6)中,芳环氟代反应的温度为50~65℃。
在所述的投料关系及反应温度下,作为优选,芳环氟代反应的时间为3-5h。
芳环氟代反应结束后,将反应体系的温度降至室温,例如降至20~35℃;优选为25~30℃;随后再在冰浴条件下,向芳环氟代反应液中投加双氧水,进行氰基水解反应。
作为优选,步骤(6)中,3,6-二氯吡嗪-2-甲腈、H2O2的投加摩尔比为1∶3~5。
所述的双氧水例如为浓度30%H2O2水溶液。
步骤(6)中,氰基水解反应的反应温度为20~35℃;优选为25~30℃。
步骤(6)中,氰基水解反应的反应时间为1~3h。
步骤(6)中,氰基水解反应结束后,直接向氰基水解反应液中投加碱水溶液,进行芳环羟基取代反应。
所述的碱水溶液优选为弱碱的水溶液,进一步优选为水溶性碳酸盐或碳酸氢盐的水溶液。
所述的碳酸氢盐优选为碳酸氢钠。
作为优选,步骤(6)中,氰基水解反应结束后,向反应液中投加水和碳酸氢钠,进行芳环羟基取代反应。
作为优选,步骤(6)中,3,6-二氯吡嗪-2-甲腈与碳酸氢钠的摩尔比为1∶0.3~0.6。
作为优选,步骤(6)中,芳环羟基取代反应的温度为40~60℃。
作为优选,步骤(6)中,芳环羟基取代反应的7~9h。
芳环羟基取代反应时间为7~8.5h,产物收率和纯度较高,超出8.5h,副反应增多,收率会有较大的下降。
步骤(6)中,芳环羟基取代反应结束后,采用酸液调节反应体系的pH至酸性,例如调整值pH为0.5~1.5;随后采用EA萃取、萃取有机相采用饱和食盐水洗涤、无水硫酸钠干燥、浓缩,制得目的产物的粗品。
本发明中,采用重结晶的方法对目的产物的粗品进行结晶。
作为优选,步骤(6)中,芳环羟基取代反应得到的法匹拉韦粗品经重结晶纯化。
进一步优选,步骤(6)中,重结晶的溶剂为甲醇、乙醇、正丙醇、异丙醇中的至少一种,进一步优化为乙醇。
本发明优选的法匹拉韦的合成方法,包括以下步骤:
步骤(a):将3-氨基吡嗪-2-羧酸(1)中加入甲醇,冰水浴下加入浓硫酸,浓硫酸滴加时间为0.5~3h,其中3-氨基吡嗪-2-羧酸(1)、甲醇和浓硫酸的重量比为1∶6~9∶1~3,在20~35℃下搅拌反应36~60h,TLC监控反应,待反应完全,浓缩,饱和碳酸钠调pH=7~8,抽滤,干燥得棕色固体3-氨基-2-酯基吡嗪(2);
步骤(b):将步骤(a)制得的物质加入乙腈,室温搅拌,分批加入NBS,其中步骤(a)所得的物质NBS的摩尔量比为1∶1.0~1.1,与乙腈的重量比为1∶7~10,25~35℃下搅拌反应,TLC监控反应,待反应完全,加入水,用Na2CO3溶液调节pH=7~8,乙酸乙酯萃取,合并有机相,经无水硫酸钠干燥后过滤,减压蒸除溶剂,得到3-氨基-6-溴吡嗪-2-羧酸甲酯粗品,将粗品溶于二氯甲烷中加热回流,抽滤除去部分杂质,浓缩,其中粗品与二氯甲烷的重量比为1∶25~50,将浓缩物溶于乙醇中重结晶,浓缩物与乙醇的重量比为1∶5~30,析晶温度为25~30℃,所得晶体经干燥,得到浅黄色固体3-氨基-6-溴吡嗪-2-羧酸甲酯(3);
步骤(c):将上述3加入浓硫酸,0℃下分批加入亚硝酸钠后,其中,物质3、浓硫酸和亚硝酸钠的摩尔比为1∶15~20∶1.5~2.5,升到室温搅拌反应2h,固体几乎全部溶解,反应液缓慢倒入冰水中,搅拌反应1h,乙酸乙酯萃取,无水硫酸钠干燥,过滤浓缩,得黄色固体3-羟基-6-溴吡嗪-2-羧酸甲酯(4);
步骤(d):将上述4加入氨水,其中,4与氨水的重量比为1∶8~15,室温搅拌反应3h,TLC板观察原料完全反应,将反应液中加入水中,乙酸乙酯萃取,有机相用无水硫酸钠干燥,减压浓缩得黄色固体3-羟基-6-溴吡嗪-2-酰胺(5);
步骤(e):在上述物质中5中加入POCl3,加热至70℃搅拌使其成均相溶液,降至室温,逐滴加入DIEA,其中5、POCl3和DIEA的摩尔量比为1∶3~5∶2~4,60℃搅拌反应1h,80℃反应1h,100℃反应4h,之后倒入冰水中剧烈搅拌反应2h,抽滤得到棕色固体,用石油醚重结晶得3,6-二氯吡嗪-2-甲腈(6),其中棕色固体与石油醚的重量比为1∶10~30;
步骤(f):包括以下的串联一锅步骤:
步骤(f-1):KF与TBAB溶于甲苯与二甲基亚砜的混合溶剂中,化合物6、KF和TBAB摩尔量比为1∶5~7∶0.05~0.5,化合物6与二甲基亚砜的重量比为1∶10~15,旋蒸两次,甲苯共沸除去水,加入化合物6,50℃下搅拌3h;
步骤(-2):冰浴下加入30%H2O2,然后在27℃下反应2h,其中化合物6与30%H2O2的摩尔量比为1∶3~5;
步骤(f-3):加入水和NaHCO3,其中化合物6与NaHCO3的摩尔量比为1∶0.3~0.6,升至50℃反应8h,然后冰浴下用6N HCl,调节pH=1.0,乙酸乙酯萃取,有机层用饱和食盐水洗两次,无水硫酸钠干燥,蒸除溶剂,得7粗品,乙醇重结晶,50℃真空干燥2h,得到类白色固体的法匹拉韦7。
步骤f的总收率为65%(由反应物6的量计算得到)。制得的法匹拉韦的mp:175-177℃。
有益效果
第一,这是一条新的法匹拉韦制备路线;本发明独创性地采用步骤(6)顺次进行的芳环氟代反应、双氧水氰基水解反应和芳环羟基取代反应的串联合成思路,可明显简化制备流程,还可明显控制目标产物的杂质含量和收率;通过研究发现,步骤(6)的收率可高达65%及以上,相较于现有的制备方法,收率提升50%以上。
第二,本发明采用所述的重结晶方法对合成线路的关键中间体3和6进行纯化,相较于现有的色谱纯化方式,本发明方法操作更简便,产物的收率更高;尤其适合于工业放大生产;且通过本发明所述的结晶方法,可以良好的控制关键中间体中的杂质含量,进而利于提升最终目的产物的质量。
第三,最后三步反应经实验发现,反应溶剂相同,故而可一步完成,提高了收率,对反应设备要求低,绿色环保;
第四,对反应中间体的后处理方法均为重结晶法,有利于工业化生产。
第五,提高了整个反应的总收率,达到26%。
附图说明
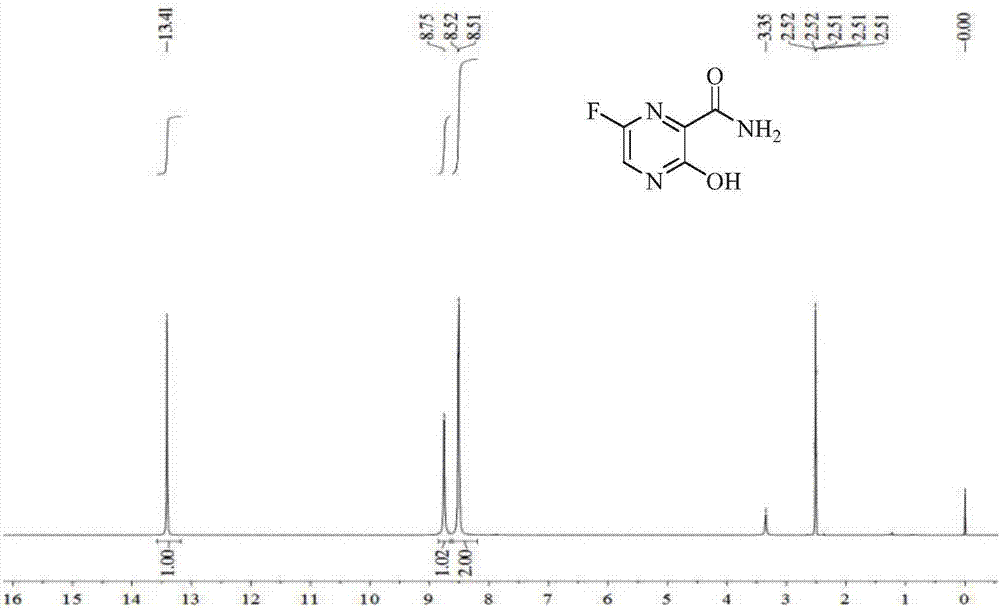
图1为法匹拉韦的19F NMR谱图;
图2为法匹拉韦的1H NMR谱图;
图3为法匹拉韦的13C NMR谱图;
图4为实施例1的6-2羟基取代反应时间对生成7收率的关系图;
具体实施方式
实施例1
第一步:制备3-氨基-2-酯基吡嗪(2)
化合物1(5g,leq)加入甲醇(50ml),冰水浴下加入浓硫酸(4eq),室温搅拌反应,TLC显示反应完全,浓缩,饱和碳酸钠调pH=8,抽滤,50℃干燥2h,得棕色固体2(4.18g,76%)。
第二步:制备3-氨基-6-溴吡嗪-2-羧酸甲酯(3)
化合物2(27.6g,leq)加入乙腈(276ml),室温搅拌,分批加入NBS(25.1g,1.01eq),室温搅拌过夜,TLC显示反应完后(20~30h),加入水(300ml),用Na2CO3溶液调节pH=7,乙酸乙酯萃取(3×50ml),合并有机相,经无水硫酸钠干燥后过滤,减压蒸除溶剂,得到3-氨基-6-溴吡嗪-2-羧酸甲酯粗品,加入30倍的二氯甲烷(所得粗品与二氯甲烷的重量比为1∶25~50)回流0.5h后,抽滤,将母液蒸除二氯甲烷,再用20倍的乙醇(旋蒸物与乙醇的重量比为1∶5~30)重结晶,在25~30℃下析晶得到浅黄色固体3,收率89%。
第三步:制备3-羟基-6-溴吡嗪-2-羧酸甲酯(4)
化合物3(39.47g,leq)加入浓硫酸(158ml),-5-0℃分批加入亚硝酸钠(23.68g,2eq),升到室温搅拌反应2h,将反应液缓慢倒入冰水中,反应1h,乙酸乙酯萃取,无水硫酸钠干燥,过滤,母液浓缩得黄色固体4,收率90%。
第四步:制备6-溴-3-羟基吡嗪-2-酰胺(5)
化合物4(40g,leq)加入氨水(400ml),室温搅拌反应3h,TLC观察4反应完全,剩余物中加入水(500ml),乙酸乙酯萃取(3×100ml),合并有机相,无水硫酸钠干燥,减压浓缩得黄色固体5,收率93.6%。
第五步:制备3,6-二氯吡嗪-2-甲腈(6)
化合物5(2g,1eq)加入POCl3(5.6g,4eq),加热至70℃,使其成均相溶液,降至室温,逐滴加入DIEA(3.57g,3eq),60℃搅拌反应1h,80℃反应1h,100℃反应4h,之后倒入冰水(110ml)中剧烈搅拌反应2h,抽滤,滤饼用20倍的石油醚(所得粗品与石油醚的重量比为1∶10~30)重结晶,得到棕色固体6收率70%。
第六步:制备3,6-二氟吡嗪-2-甲酰胺(7)
KF(1g,6eq)与TBAB(372.3mg,0.4eq)溶于甲苯(10ml)与二甲基亚砜(5ml)的混合溶剂中,共沸除去水后,加入化合物6(500mg,1eq),55℃条件下,搅拌3h。TLC显示原料反应完全后,降至室温后,冰浴下加入30%H2O2(0.35ml),在27℃下反应2h,加入水(1ml)和NaHCO3(0.132g,1.57mmol),50℃反应8.5h,冰浴下用6N HCl,调节pH=1.0,乙酸乙酯(4×5ml)萃取,有机层用饱和食盐水洗两次,无水硫酸钠干燥,蒸除有机溶剂,用20倍的乙醇重结晶(粗品与乙醇的重量比为1∶5~30),得类白色固体7(法匹拉韦),收率为65%(由反应物6的量计算得到)。mp:175-177℃。
法匹拉韦的19F NMR(376MHZ)谱图见图1;
法匹拉韦的1HNMR(500MHz)谱图见图2;
法匹拉韦的13C NMR(126MHz)谱图见图3;
图4为实施例1的6-2羟基取代反应时间对生成7收率的关系图;
实施例1中,第六步中,投加NaHCO3进行芳基羟基化反应的时间与收率的关系见图4所示。
由图4可知:当反应时间低于8.5h时,随着反应时间的延长,收率有所提高,因为此时间段内原料没有反应完全,故而,温度升高,收率有所改变;当反应时间超过8.5h后,副反应增多,收率会有较大的下降。
实施例2
和实施例1相比,区别仅在于:
第五步:制备3,6-二氯吡嗪-2-甲腈(6)
将化合物5(2g,1eq)溶于氯苯(10ml)中,缓慢滴加POCl3(5.6g,4eq),加热至70℃,使其成均相溶液,降至室温,逐滴加入DIEA(3.57g,3eq),60℃搅拌反应1h,80℃反应1h,100℃反应4h,之后倒入冰水(110ml)中剧烈搅拌反应2h,抽滤,滤饼用20倍的石油醚(所得粗品与石油醚的重量比为1∶10~30),得到棕色固体6,收率60%。
其余步骤与实施案例1相同。
实施例3
实施例1相比,区别仅在于:
将第二步得到3-氨基-6-溴吡嗪-2-羧酸甲酯粗品,加入30倍的二氯甲烷(所得粗品与二氯甲烷的重量比为1∶25~50)回流0.5h后,抽滤,将母液蒸除二氯甲烷,再用20倍的甲醇(旋蒸物与甲醇的重量比为1∶5~30)重结晶,得到浅黄色固体3,收率76%。
对比例1
实施例1相比,区别仅在于:
将第二步得到3-氨基-6-溴吡嗪-2-羧酸甲酯粗品,用20倍的二氯甲烷(粗品与二氯甲烷的重量比为1∶5~30)重结晶。
对比例2
实施例1相比,区别仅在于:
将第二步得到3-氨基-6-溴吡嗪-2-羧酸甲酯粗品,用20倍的EA(粗品与EA的重量比为1∶5~30)重结晶。
对比例3
实施例1相比,区别仅在于:
将第二步得到3-氨基-6-溴吡嗪-2-羧酸甲酯粗品,用20倍的甲醇(粗品与甲醇的重量比为1∶5~30)重结晶。
对比例4
实施例1相比,区别仅在于:
将第二步得到3-氨基-6-溴吡嗪-2-羧酸甲酯粗品,用20倍的乙醇(粗品与甲醇的重量比为1∶5~30)重结晶。
对比例5
实施例1相比,区别仅在于:
将第二步得到3-氨基-6-溴吡嗪-2-羧酸甲酯粗品,用20倍的二氯甲烷/石油醚混合溶剂(体积比1∶1)(粗品与该混合溶剂的重量比为1∶5~30)重结晶。
对比例6
实施例1相比,区别仅在于:
将第二步得到3-氨基-6-溴吡嗪-2-羧酸甲酯粗品,用20倍的乙酸乙酯/石油醚混合溶剂(体积比1∶1)(粗品与该混合溶剂的重量比为1∶5~30)重结晶。
以6g实施例1制得的3-氨基-6-溴吡嗪-2-羧酸甲酯粗品为原料,分别采用实施例1、实施例3和对比例1~6的结晶方法进行结晶处理,结晶结果如表1所示:
表1
由表1可知,对于3-氨基-6-溴吡嗪-2-羧酸甲酯的重结晶,采用单一溶剂:二氯甲烷、乙酸乙酯、乙醇、甲醇等进行重结晶过程,发现多次重结晶之后仍得不到纯净的3-氨基-6-溴吡嗪-2-羧酸甲酯;采用多种混合溶剂二氯甲烷/石油醚、乙酸乙酯/石油醚等进行此过程,仍然得不到纯品;在重结晶摸索过程中,预先经二氯甲烷除杂处理,再用单一溶剂结晶,通过实验发现使用甲醇或乙醇可得纯品;乙醇效果较好。
实施例4
实施例1相比,区别仅在于:
将第五步所得到的3,6-二氯吡嗪-2-甲腈(6)粗品,加入20倍的甲醇(所得粗品与甲醇的重量比为1∶10~30),加热回流0.5h,抽滤,除去不溶物,在25-30℃下析晶,得到浅黄色固体6,收率为30%。
实施例5
实施例1相比,区别仅在于:
将第五步所得到的3,6-二氯吡嗪-2-甲腈(6)粗品,加入20倍的二氯甲烷(所得粗品与二氯甲烷的重量比为1∶10~30),加热回流0.5h,抽滤,除去不溶物,在25-30℃下析晶,得到浅黄色固体6,收率为40%。
对比例7
和实施例1相比,区别仅在于:
将氟代产物在冰浴下加入醋酸293mg、三乙胺494mg,在27℃下搅拌反应2h,冰浴下加入30%H2O2(0.35ml),在27℃下反应2h,乙酸乙酯(4×5ml)萃取,有机层用饱和食盐水洗两次,无水硫酸钠干燥,蒸除有机溶剂,得粗品,用20倍的乙醇(所得粗品与乙醇的重量比为1∶5~30)重结晶,得类白色固体7,收率为42%(由反应物6的量计算得到)。
对比例8
和实施例1相比,区别仅在于:
将氟代产物在冰浴下加入醋酸293mg、三乙胺494mg,在27℃下搅拌反应2h,加入浓硫酸(1.5ml),50℃反应4h,缓慢滴入到冰水中,搅拌反应1h,乙酸乙酯(4×5ml)萃取,有机层用饱和食盐水洗两次,无水硫酸钠干燥,蒸除有机溶剂,得粗品,用20倍的乙醇(所得粗品与乙醇的重量比为1∶5~30)重结晶,得类白色固体7,收率为41%(由反应物6的量计算得到)。
对比例9
和实施例1相比,区别仅在于:
将氟代产物在冰浴下加入水(1ml)和NaHCO3(0.132g,1.57mmol),50℃反应8.5h,冰浴下加入30%H2O2(0.35ml),在27℃下反应2h,冰浴下用6N HCl,调节pH=1.0,乙酸乙酯(4×5ml)萃取,有机层用饱和食盐水洗两次,无水硫酸钠干燥,蒸除有机溶剂,得粗品,用20倍的乙醇(所得粗品与乙醇的重量比为1∶5~30)重结晶,得类白色固体7,收率为54%(由反应物6的量计算得到)。mp:175-177℃。
以实施例1制得的氟代产物为原料,分别采用对比例7~9合成目的产物,合成线路见以下方程式,经过串联的a、b、c步骤;
实施例1、对比例7~9合成的检测数据见表2
表2
由表2可知:与实施案例(1)相比,区别在于氟代反应、腈水解反应、羟基取代反应的顺序,由表2可知常温下在醋酸和三乙胺的催化下,羟基取代反应的转化率很低,所以优化选为在碱水溶液中进行;而在浓硫酸催化下进行的腈水解反应复杂,副产物多,分离提纯困难;如果先进行在碳酸氢钠催化下的羟基取代反应,在进行腈水解反应,副产物较多,同样分离提纯困难。故而,本发明独创的过氧化氢催化下进行腈水解反应,配合所述的依次进行氟代反应;腈基水解反应;羟基取代反应,在此优化条件下副反应少,分离提纯容易,有利于工业化生产。
- 还没有人留言评论。精彩留言会获得点赞!