一种高纯度卡利拉嗪的精制方法与流程
本发明属于药物化学领域,具体涉及一种高纯度卡利拉嗪的精制方法。
背景技术:
盐酸卡利拉嗪,化学名为反式-1-{4-[2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基]-环己基}-3,3-二甲基脲盐酸盐,是一种d2和d3受体部分激动剂,特别对d3受体有较高的选择性,对5-ht1a也有部分激动作用,由gedeonrichter和forestlaboratories公司共同开发。2015年9月17日,fda批准其用于治疗精神分裂症和双相情感障碍。
卡利拉嗪结构如下所示:
目前国内外公开的卡利拉嗪的制备方法主要有2类:
(1)专利cn1829703报道了以2,3-二氯苯基哌嗪(vii)和反式叔丁基(4-(2-氧代乙基)环己基)甲酸铵(x)为关键中间体,经过氨化还原、脱保护得到中间体ix。最后酰化中间体ix,得到卡利拉嗪。
(2)专利cn102256953报道了以反式氨基环己基乙酸乙酯为起始原料,经过氨基保护、还原、磺酰化反应制备关键中间体xi;xi再与2,3-二氯苯基哌嗪盐酸盐(vii)经过缩合、脱保护得到中间体ix。最后酰化中间体ix,得到卡利拉嗪。
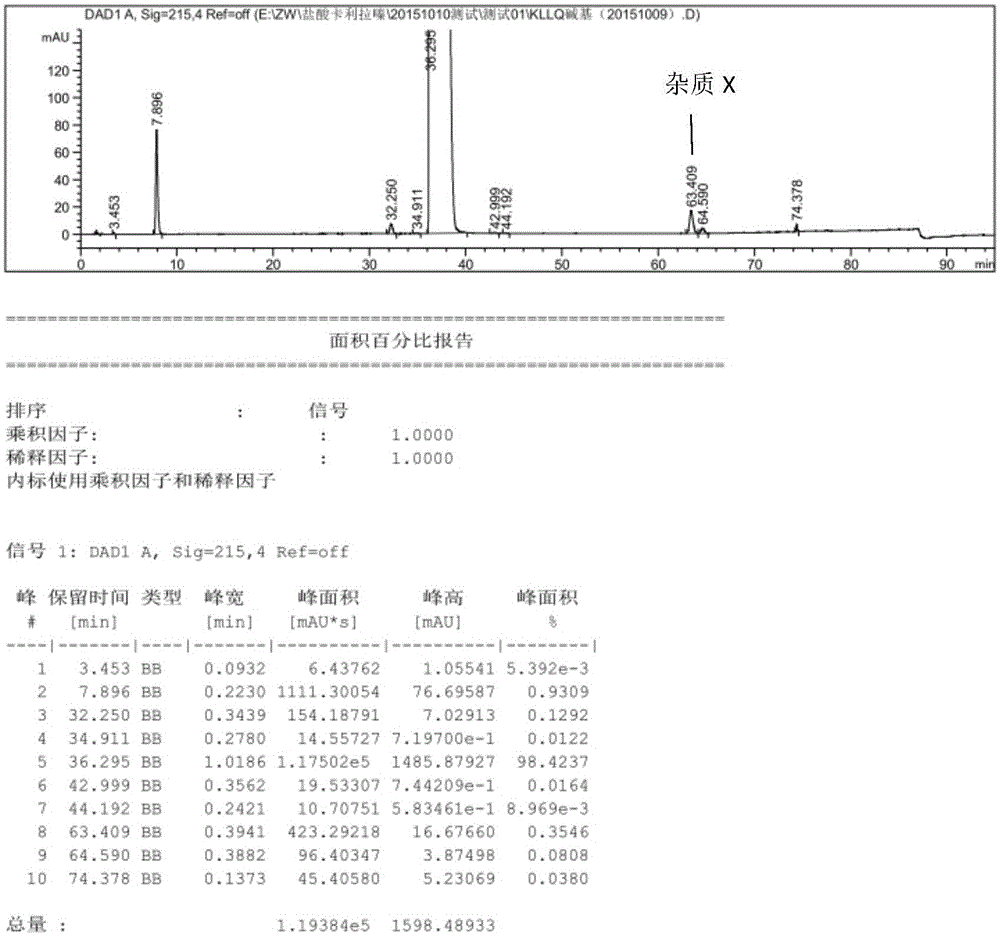
在制备卡利拉嗪过程中,研发人员发现产品纯度不高,在卡利拉嗪主峰前面出现一个较大的杂质峰,很难除去,经富集,结构确证,确定为杂质x,结构如下:
杂质x系首次提纯发现,在成盐过程中,该杂质并不能有效去除,会在盐酸卡利拉嗪中富集,使卡利拉嗪终产品不合格,需要经过多次重结晶进行去除,进而影响卡利拉嗪收率,且耗时较长。
而现有技术中没有报道过卡利拉嗪的精制方法,为了解决获得高纯度卡利拉嗪的方法,需要提供一种易于工业化生产,且能高效去除副产物(x)的方法。
技术实现要素:
:
本发明为了解决现有技术中的副产物x不易去除的问题,提供一种卡利拉嗪的精制方法,使杂质在卡利拉嗪成盐之前进行有效去除,提高了产品纯度。
本发明提供如下技术方案来解决现有技术问题:
本发明提供一种卡利拉嗪的精制方法,其特征在于,该精制方法包括以下步骤:
1)将卡利拉嗪粗品加入二氯甲烷和c1-5的醇,加热搅拌至溶解;
2)加入酯类溶剂或者酮类溶剂,降温析晶;
3)过滤收集卡利拉嗪晶体,用乙酸乙酯洗涤,真空干燥得到高纯度的卡利拉嗪。
进一步的,在上述精制方法中,所述的步骤1中c1-5的醇为甲醇、乙醇、异丙醇中的一种或两种以上混合物。
进一步的,在上述精制方法中,所述的步骤1中酯类溶剂为乙酸乙酯、乙酸甲酯中的一种或两种混合物;酮类溶剂为丙酮或者丁酮中的一种或两种混合物;所述的酯类溶剂为乙酸乙酯,酮类溶剂为丙酮。
进一步的,在上述精制方法中,所述的投料比为卡利拉嗪:二氯甲烷:c1-5的醇:酯类溶剂或者酮类溶剂为1g:1~30ml:1~30ml:5~100ml。所述的投料比优选为卡利拉嗪:二氯甲烷:c1-5的醇:乙酸乙酯(或丙酮)为1g:1~30ml:1~30ml:5~100ml。所述的投料比优选为卡利拉嗪:二氯甲烷:乙醇:乙酸乙酯(或丙酮)为1g:2~10ml:3~10ml:5~15mll。
所述的投料比优选为卡利拉嗪:二氯甲烷:乙醇:乙酸乙酯(或丙酮)为1g:5~10ml:3~10ml:5~15ml。
进一步的,在上述精制方法中,,所述的析晶温度为0~30℃,析晶时间为1~5小时。
进一步的,在上述精制方法中,所述的析晶温度为0~10℃,析晶时间为2~3小时。
本发明的有益技术效果:
本发明解决现有技术中的副产物x不易去除的问题,提供一种卡利拉嗪的精制方法,使杂质在卡利拉嗪成盐之前进行有效去除,提高了产品纯度,有利于提高产品质量,且该精制方法简单,所用试剂环保,易于工业化生产的使用。
附图说明:
图1对比实施例1中专利路线制备的卡利拉嗪高效液相色谱图
图2实例1制备的卡利拉嗪高效液相色谱图
图3实例2制备的卡利拉嗪高效液相色谱图
图4实例3制备的卡利拉嗪高效液相色谱图
图5实例4制备的卡利拉嗪高效液相色谱图
对比实施例1:
试验人员用如下方法制备卡利拉嗪:
向5l四口反应瓶中加入3500ml二氯甲烷、500ml的40%氢氧化钠水溶液、10g的四丁基溴化铵和62g的n,n-二甲基氨基甲酰氯。室温搅拌30分钟;加入130g反式4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己胺二盐酸盐,氮气氛下,加热到50℃,剧烈回流20小时。反应完毕,冷却到室温,萃取分离,有机相分别用3x1500ml水和1500ml饱和食盐水洗涤。无水硫酸钠干燥,过滤;浓缩除去溶剂,余下少量体积,室温搅拌2小时,过滤,并于50℃真空干燥至恒重,得到120g类白色固体。hplc含量为98.42%,杂质x(63.409min)含量为0.345%。
实施例1、
(1)将卡利拉嗪粗品10g加入到250ml反应瓶中,加入50ml二氯甲烷、30ml乙醇,加热回流,使得溶液澄清;
(2)缓慢加入75ml乙酸乙酯,搅拌20分钟;缓慢降温到30℃。
(3)冰水浴,冷却到0-10℃,搅拌3小时;
(4)过滤,并用30ml乙酸乙酯洗涤滤饼,并于50℃真空干燥至恒重,得到8.1g卡利拉嗪晶体,收率81%,纯度(hplc)99.79,杂质x(63.78min)的含量0.02%,如图2所述。
实施例2、
(1)将卡利拉嗪粗品10g加入到250ml反应瓶中,加入30ml二氯甲烷、70ml乙醇,加热回流,使得溶液澄清;
(2)缓慢加入75ml乙酸乙酯,搅拌20分钟;缓慢降温到30℃。
(3)冰水浴,冷却到0-10℃,搅拌5小时;
(4)过滤,并用15ml乙酸乙酯洗涤滤饼,并于50℃真空干燥至恒重,得到8.4g卡利拉嗪晶体,收率84%,纯度(hplc)99.68,杂质x(63.33min)的含量0.04%,如图3所示。
实施例3、
(1)将卡利拉嗪粗品20g加入到500ml反应瓶中,加入40ml二氯甲烷、60ml乙醇,加热回流,使得溶液澄清;
(2)缓慢加入100ml乙酸乙酯,搅拌20分钟;缓慢降温到30℃。
(3)冰水浴,冷却到0-10℃,搅拌3小时;
(4)过滤,并用30ml乙酸乙酯洗涤滤饼,并于50℃真空干燥至恒重,得到17.1g卡利拉嗪晶体,收率85.5%,纯度(hplc)99.88,杂质x的含量0.02%,如图4所示。
实施例4、
(1)将卡利拉嗪粗品20g加入到500ml反应瓶中,加入100ml二氯甲烷、120ml乙醇,加热回流,使得溶液澄清;
(2)缓慢加入100ml丙酮,搅拌20分钟;缓慢降温到30℃。
(3)冰水浴,冷却到0-10℃,搅拌3小时;
(4)过滤,并用30ml丙酮洗涤滤饼,并于50℃真空干燥至恒重,得到16.0g卡利拉嗪晶体,收率80%,纯度(hplc)99.82,杂质x的含量0.02%,如图5所示。
实施例5、
(1)将卡利拉嗪粗品10g加入到250ml反应瓶中,加入50ml二氯甲烷、30ml乙醇,加热回流,使得溶液澄清;
(2)缓慢加入60ml丁酮,搅拌20分钟;缓慢降温到30℃。
(3)冰水浴,冷却到0-10℃,搅拌3小时;
(4)过滤,并用20ml丁酮洗涤滤饼,并于50℃真空干燥至恒重,得到8.2g卡利拉嗪晶体,收率82%,纯度(hplc)99.56%,杂质x的含量0.03%。
实施例6、
(1)将卡利拉嗪粗品10g加入到250ml反应瓶中,加入50ml二氯甲烷、30ml乙醇,加热回流,使得溶液澄清;
(2)缓慢加入75ml乙酸乙酯,搅拌20分钟;缓慢降温到30℃。
(3)冰水浴,冷却到0-10℃,搅拌3小时;
(4)过滤,并用15ml乙酸乙酯洗涤滤饼,并于50℃真空干燥至恒重,得到8.1g卡利拉嗪晶体,收率83%,纯度(hplc)99.57%,杂质x的含量0.02%。
实施例7、
(1)将卡利拉嗪粗品10g加入到250ml反应瓶中,加入70ml二氯甲烷、30ml甲醇,加热回流,使得溶液澄清;
(2)缓慢加入75ml乙酸乙酯,搅拌20分钟;缓慢降温到30℃。
(3)冰水浴,冷却到0-10℃,搅拌3小时;
(4)过滤,并用15ml乙酸乙酯洗涤滤饼,并于50℃真空干燥至恒重,得到8.3g卡利拉嗪晶体,收率83%。
实施例8、
(1)将卡利拉嗪粗品10g加入到250ml反应瓶中,加入50ml二氯甲烷、15ml甲醇,加热回流,使得溶液澄清;
(2)缓慢加入75ml乙酸乙酯,搅拌20分钟;缓慢降温到30℃。
(3)冰水浴,冷却到0-10℃,搅拌3小时;
(4)过滤,并用15ml乙酸乙酯洗涤滤饼,并于50℃真空干燥至恒重,得到8.4g卡利拉嗪晶体,收率86%,纯度(hplc)99.51%,杂质x的含量0.03%。
实施例9、
(1)将卡利拉嗪粗品10g加入到250ml反应瓶中,加入20ml二氯甲烷、30ml甲醇,加热回流,使得溶液澄清;
(2)缓慢加入50ml乙酸乙酯,搅拌20分钟;缓慢降温到30℃。
(3)冰水浴,冷却到0-10℃,搅拌3小时;
(4)过滤,并用15ml乙酸乙酯洗涤滤饼,并于50℃真空干燥至恒重,得到8.4g卡利拉嗪晶体,收率84%。
实施例10、
(1)将卡利拉嗪粗品10g加入到500ml反应瓶中,加入100ml二氯甲烷、100ml甲醇,加热回流,使得溶液澄清;
(2)缓慢加入50ml乙酸乙酯,搅拌20分钟;缓慢降温到30℃。
(3)冰水浴,冷却到0-10℃,搅拌3小时;
(4)过滤,并用15ml乙酸乙酯洗涤滤饼,并于50℃真空干燥至恒重,得到8.4g卡利拉嗪晶体,收率84.6%。
实施例11、
(1)将卡利拉嗪粗品10g加入到500ml反应瓶中,加入50ml二氯甲烷、40ml甲醇,加热回流,使得溶液澄清;
(2)缓慢加入150ml乙酸乙酯,搅拌20分钟;缓慢降温到30℃。
(3)冰水浴,冷却到0-10℃,搅拌3小时;
(4)过滤,并用15ml乙酸乙酯洗涤滤饼,并于50℃真空干燥至恒重,得到8.4g卡利拉嗪晶体,收率82%。
实施例12、
(1)将卡利拉嗪粗品1g加入到250ml反应瓶中,加入30ml二氯甲烷、20ml甲醇,加热回流,使得溶液澄清;
(2)缓慢加入100ml乙酸乙酯,搅拌20分钟;缓慢降温到30℃。
(3)冰水浴,冷却到0-10℃,搅拌3小时;
(4)过滤,并用15ml乙酸乙酯洗涤滤饼,并于50℃真空干燥至恒重,得到8.4g卡利拉嗪晶体,收率83%。
通过上述实施例以及附图1-5,可以看出该方法可以显著减低杂质x的含量,且方法简单易于工业化生产。
- 还没有人留言评论。精彩留言会获得点赞!