吡咯并吡唑类衍生物、其制备方法及其在医药上的应用与流程
[0001]
本发明涉及一类新的吡咯并吡唑类衍生物、其制备方法以及其本身或含有此类衍生物的药物组合物作为治疗剂特别是作为胃酸分泌抑制剂和钾离子竞争性酸阻滞剂(p-cabs)的用途。
背景技术:
[0002]
消化性溃疡主要指发生于胃和十二指肠的慢性溃疡。虽然有地区差异,但消化性溃疡发病率通常占总人口的10%~20%,是一多发病、常见病。溃疡的形成有各种因素,其中酸性胃液对黏膜的消化作用是溃疡形成的基本因素。因此,抑制胃酸分泌逐渐成为治疗消化性溃疡类疾病的首选方法。
[0003]
自1988年第一个质子泵抑制剂(proton pump inhibitors,ppis)奥美拉唑上市以来,至今全球已有数个ppis产品上市,包括兰索拉唑、泮托拉唑、雷贝拉唑和艾司奥美拉唑等。ppis已经成为治疗胃酸相关性疾病包括消化性溃疡、反流性食管炎和卓-艾综合征等疾病的首选药物。质子泵(proton pump)实质为h
+
/k
+-腺苷三磷酸酶(h
+
/k
+-atpase),它特异性的将质子(h+)泵入胃腔从而形成胃内的强酸性。质子泵抑制剂可以抑制质子泵的活性从而调节质子泵介导的胃酸分泌。
[0004]
钾离子竞争性酸阻滞剂(potassium-competitive acid blockers,p-cabs)是一类新型的胃酸阻滞剂,它通过可逆的、与钾离子(k
+
)竞争性的结合h
+
/k
+-atpase从而起到抑制h
+
/k
+-atpase酶活性的作用。与ppis相比较,p-cabs具有亲脂性、弱碱性以及在酸性(低ph)条件下稳定等特点。同时,p-cabs具有起效迅速以及比较容易达到抑酸效果等优势。
[0005]
第一款p-cabs新药沃诺拉赞于2014年在日本上市,用于治疗胃酸相关疾病如消化性溃疡。一系列的钾离子竞争性酸阻滞剂结构也已经被公开。但仍然需要开发结构类型多样性的具有更好成药性的新化合物。
技术实现要素:
[0006]
针对上述问题,本发明的目的在于提供新的结构类型且具有优异的效果和作用的用于治疗胃酸相关疾病如消化性溃疡的化合物。
[0007]
一方面,本发明提供一种通式(i)所示的化合物或其药学上可接受的盐,其中:
n=1或2;r1选自氢原子、卤素或烷基;r2选自氢原子、卤素、羟基或烷基;r3、r4各自独立选自氢原子、任选地被取代的烷基或任选地被取代的环烷基。
[0008]
优选地,n=1或2;r1为卤素;r2选自氢原子或卤素;r3、r4各自独立选自氢原子、任选地被取代的c
1~3
烷基或任选地被取代的三元、四元、五元或六元环烷基。
[0009]
优选地,n=1或2;r1为卤素;r2选自氢原子或卤素;r3、r4各自独立选自氢原子、任选地被一个或多个羟基、氟原子或氨基取代的c
1~3
烷基或任选地被一个或多个羟基、氟原子或氨基取代的三元、四元、五元或六元环烷基。
[0010]
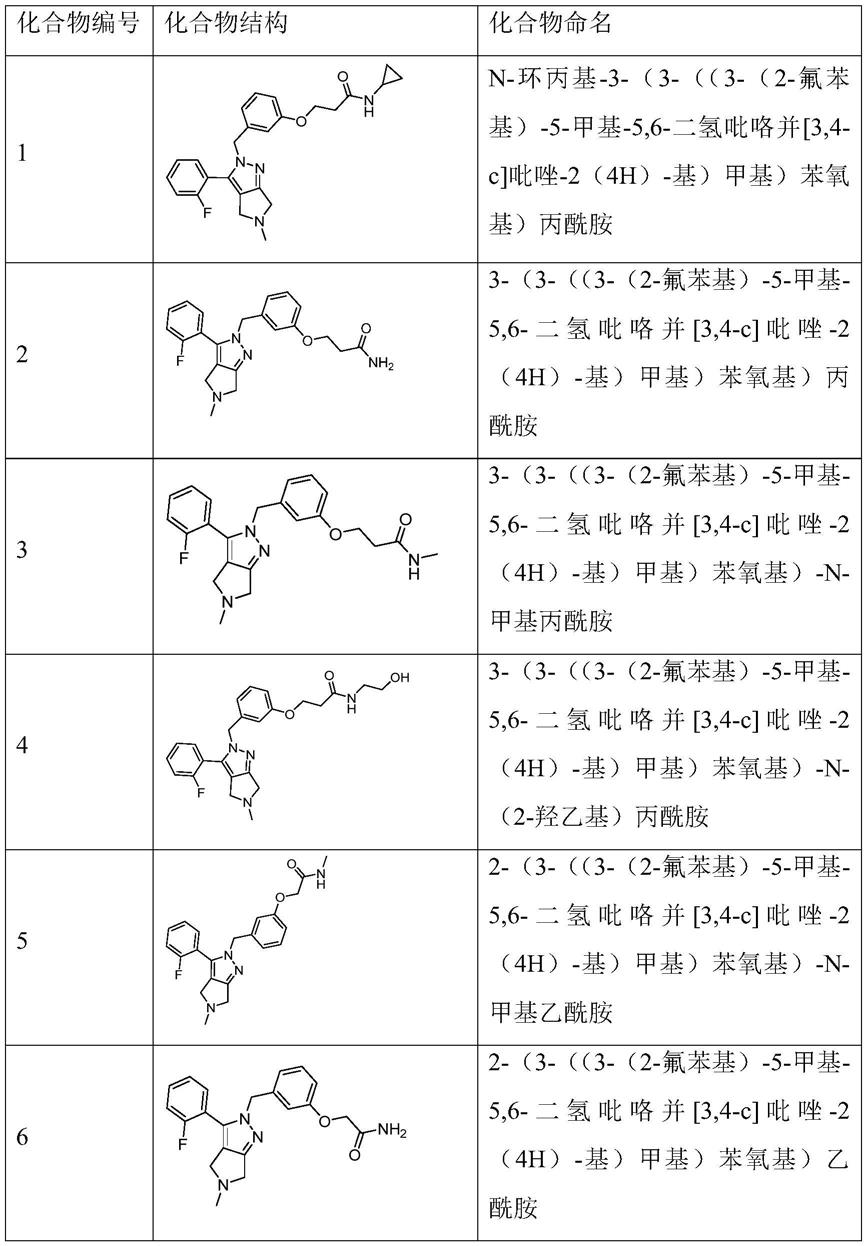
优选地,n=1或2;r1为氟原子或氯原子;r2选自氢原子或氟原子;r3、r4各自独立选自氢原子、任选地被一个羟基取代的c
1~3
烷基或环丙基。
[0011]
优选地,所述化合物选自:n-环丙基-3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)丙酰胺;3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)丙酰胺;3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-甲基丙酰胺;3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-(2-羟乙基)丙酰胺;2-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-甲基乙酰胺;2-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)乙酰胺;n-环丙基-2-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)乙酰胺;3-(3-((3-(2-氯苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-(2-羟乙基)丙酰胺。
[0012]
第二方面,本发明提供一种药物组合物,包括通式(i)所示的化合物或其药学上可接受的盐以及药学上可接受的载体、赋形剂或稀释剂。
[0013]
第三方面,本发明提供通式(i)所示的化合物或其药学上可接受的盐或上述药物组合物在制备胃酸分泌抑制剂中的用途。
[0014]
第四方面,本发明提供通式(i)所示的化合物或其药学上可接受的盐或上述药物组合物在制备h
+
/k
+-腺苷三磷酸酶(h
+
/k
+-atpase)抑制剂中的用途。
[0015]
第五方面,本发明提供通式(i)所示的化合物或其药学上可接受的盐或上述药物组合物在制备钾离子竞争性酸阻滞剂(p-cabs)中的用途。
[0016]
第六方面,本发明提供通式(i)所示的化合物或其药学上可接受的盐或上述药物组合物在制备药物中的用途,所述药物用于治疗和/或预防消化性溃疡、卓-艾综合征、胃炎、糜烂性食管炎、反流性食管炎、症状性胃食管反流疾病、巴雷特食管炎、功能性消化不良、幽门螺旋杆菌感染、胃癌、胃malt淋巴瘤、非甾体抗炎药引起的溃疡或手术后应激导致的胃酸过多或溃疡;或者抑制消化性溃疡、急性应激性溃疡、出血性胃炎或侵入性应激造成的上消化道出血。
具体实施方式
[0017]
以下通过下述实施方式进一步说明本发明,应理解,下述实施方式仅用于说明本发明,而非限制本发明。
[0018]
除非有相反陈述,下列用在说明书和权利要求中的术语具有下述含义。
[0019]
术语“烷基”指饱和的脂族烃基团,包括1至10个碳原子的直链或支链基团。优选含有1至5个碳原子的烷基。更优选含有1至3个碳原子的烷基,例如甲基,乙基,正丙基,异丙基。
[0020]
各种含碳氢结构部分的碳原子含量由该部分的标有最小和最大碳原子数目的前缀表示,即前缀c
i~j
表示该部分的碳原子数为整数“i”至整数“j”(包括i和j)。因此,例如,c
1~3
烷基是指1至3个碳原子的烷基(包括1和3)。
[0021]
术语“环烷基”是指碳原子的环状饱和单价单环或双环碳氢基团,例如环丙基、环己基,或类似基团。
[0022]
术语“羟基”指-oh基团。
[0023]
术语“卤素”指氟、氯、溴或碘。
[0024]
术语“任选地被取代”是指取代是任选的,因此,其既包括未取代的原子和基团,又包括取代的原子和基团。“取代的”原子或基团表示所指定原子或基团上的任意氢均可被来自所指定取代基组的选择替换(直至所指定原子或基团上的每个氢原子均被来自所指定取代基组的选择替换),条件是不超过所指定原子或基团的正常化合价,并且所述取代产生稳定的化合物。例如,如果甲基(即,ch3)是任选取代的,则碳原子上的至多3个氢原子可被取代基替换。如果原子或基团被描述为任选地被一个或多个非氢取代基取代,则它可被至多所述原子或基团上的可取代位置的最大数的非氢取代基取代。
[0025]
除非特别说明,本发明中,所有出现的化合物均意在包括所有可能的异构体,例如互变异构体、对映异构体、非对映异构体、及其混合物形式。
[0026]
术语“本发明化合物”指通式(i)所示的化合物。该术语还包括通式(i)化合物的各种晶型形式、药学上可接受的盐、水合物或溶剂合物。
[0027]
术语“药学上可接受的盐”指本发明化合物与酸或碱所形成的适合用作药物的盐。药学上可接受的盐包括无机盐和有机盐。一类优选的盐是本发明化合物与酸形成的盐。适合形成盐的酸包括但不限于:盐酸、氢溴酸、氢氟酸、硫酸、硝酸、磷酸等无机酸,甲酸、乙酸、
丙酸、草酸、丙二酸、琥珀酸、富马酸、马来酸、乳酸、苹果酸、酒石酸、柠檬酸、苦味酸、甲磺酸、苯甲磺酸,苯磺酸等有机酸;以及天冬氨酸、谷氨酸等酸性氨基酸。
[0028]
术语“药学上可接受的载体”表示能用于制备药物组合物的载体,它们一般是安全的、无毒性的,不是生物上或其他方面不期待的,且包括能被动物和人类药学上接受的载体。在说明书和权利要求书中使用的“药学上可接受的载体”包括一种或一种以上的这类载体。
[0029]
术语“含有”、“包含”或“包括”表示各种成分可一起应用于本发明的混合物或组合物中。因此,术语“主要由...组成”和“由...组成”包含在术语“含有”中。
[0030]
术语“预防”例如是指在可能暴露或预先处置于疾病但尚未经历或显示疾病症状的哺乳动物中使疾病临床症状不发展。
[0031]
术语“治疗”可指抑制疾病,例如阻止或降低疾病或其临床症状的发展,或者缓解疾病,例如使疾病或其临床症状退化。
[0032]
通式(i)化合物
[0033]
本发明一些实施方式中,n=1或2。
[0034]
本发明一些实施方式中,r1选自氢原子、卤素或烷基。更优选实施方式中,r1为卤素。进一步优选实施方式中,r1为氟原子或氯原子。r1的取代位点优选为2位。
[0035]
本发明一些实施方式中,r2选自氢原子、卤素、羟基或烷基。更优选实施方式中,r2选自氢原子或卤素。进一步优选实施方式中,r2选自氢原子或氟原子。
[0036]
本发明一些实施方式中,r3、r4各自独立选自氢原子、任选地被取代的烷基或任选地被取代的环烷基。
[0037]
所述任选地被取代的烷基例如是任选地被一个或多个羟基、氟原子或氨基取代的烷基,优选为任选地被一个或多个羟基或氟原子取代的烷基,更优选为任选地被一个羟基取代的烷基。羟基优选取代烷基端位上的氢。一些实施方式中,所述任选地被取代的烷基是任选地被取代的c
1~3
烷基。
[0038]
任选地被取代的环烷基例如是任选地被一个或多个羟基、氟原子或氨基取代的环烷基,优选是任选地被一个或多个羟基或氟原子取代的环烷基。一些实施方式中,所述任选地被取代的环烷基是任选地被取代的三元、四元、五元或六元环烷基。尤其优选的实施方式中,任选地被取代的环烷基是环丙基。
[0039]
一些实施方式中,r3、r4中至少一者为氢原子。
[0040]
一些实施方式中,r3、r4中一者为氢原子,另一者选自氢原子、甲基、乙基、羟乙基、环丙基。
[0041]
本发明一些实施方式中,通式(i)化合物选自表1中所示的化合物。
[0042]
表1
[0043]
通式(i)化合物的制备方法
[0044]
本发明一些实施方式中,n=2,通式(i)化合物可用通式(ia)表示,其制备可采用如下通用合成路线1:通用合成路线1
[0045]
其中,r1、r2、r3、r4的定义如上所述。
[0046]
p1基可为本领域公知的氨基保护基,例如可选自苄基、对甲氧基苯基甲基、邻硝基苯基甲基等可被取代的c7-11芳烷基;乙酰基、三氟乙酰基等可被取代的c1-6烷羰基;苯甲酰基等可被取代的c6-10芳羰基;甲氧羰基、乙氧羰基、boc基(叔丁氧羰基)、cbz基(苄氧羰基)、fmoc基(芴甲氧羰基)、teoc基(三甲基甲硅烷基乙氧羰基)等可被取代的c1-6烷氧羰基;alloc基(烯丙氧羰基)等烯氧羰基;甲磺酰基等烷基磺酰基;对甲苯磺酰基等可被取代的c6-10芳基磺酰基。
[0047]
x1基可选自氯原子、溴原子、碘原子等卤素原子。
[0048]
x2基可为本领域公知的离去基团,例如可选自氟原子、氯原子、溴原子、碘原子等卤素原子。
[0049]
p2基可为本领域公知的羟基保护基,例如可选自酯类保护基、硅醚类保护基、烷基醚类保护基、烷氧基烷基醚类保护基等,其中优选烷氧基烷基醚类保护基,例如thp(四氢吡喃基)、mom(甲氧基甲基)、mtm(甲硫基甲基)等。
[0050]
p3基可为本领域公知的羟基保护基,例如可选自酯类保护基、硅醚类保护基、烷基醚类保护基、烷氧基烷基醚类保护基等,其中优选烷基醚类保护基,例如叔丁基等。
[0051]
步骤(a)中,将式i-1化合物与卤化试剂反应,得到式i-2化合物。卤化试剂例如可为n-碘代丁二酰亚胺、n-溴代丁二酰亚胺等。
[0052]
步骤(b)中,式i-2化合物与式i-3化合物反应,得到式i-4化合物。式i-2化合物和式i-3化合物的摩尔比可为1:(0.5~3.0)。反应溶剂可为乙腈、丙酮、四氢呋喃、二氧六环、n,n-二甲基甲酰胺等。步骤(b)的反应可在碱存在下进行。所述碱可选自:碳酸铯、碳酸钾、碳酸钠、氢氧化钾、氢氧化钠等。式i-2化合物与碱的摩尔比可为1:(1.0~6.0)。步骤(b)的反应温度可以由本领域技术人员适当设定,例如可为0~100℃。
[0053]
步骤(c)中,式i-4化合物与式i-5化合物反应,得到式i-6化合物。式i-4化合物与式i-5化合物的摩尔比可为1:(0.5~5.0)。反应溶剂可为乙二醇二甲醚、二氧六环、四氢呋喃、甲苯、n,n-二甲基甲酰胺等。步骤(c)可在钯催化剂存在下进行。所述钯催化剂可选自:四(三苯基膦)钯、[1,1
’-
双(二苯基膦基)二茂铁]二氯化钯、氯化烯丙基钯(ii)二聚物、三(二亚苄基丙酮)二钯、氯化钯等。另外,步骤(c)的反应可在碱存在下进行。所述碱可选自:碳酸钾、碳酸钠、碳酸氢钠、碳酸铯、磷酸钾等。式i-4化合物与碱的摩尔比可为1:(0.5~5.0)。步骤(c)的反应温度可以由本领域技术人员适当设定,例如可为40~150℃。
[0054]
步骤(d)中,将式i-6化合物脱除p1和p2保护基,得到式i-7化合物。反应条件优选采用本领域常用的可同时脱除氨基保护基和羟基保护基的反应条件。
[0055]
步骤(e)中,对式i-7化合物进行氨基甲基化反应,得到式i-8化合物。该步骤可采用本领域公知的氨基甲基化反应条件。一些实施方式中,将式i-7化合物与甲醛搅拌一段时间生成席夫碱,然后加入还原剂例如醋酸硼氢化钠反应一段时间,得到式i-8化合物。
[0056]
步骤(f)中,将式i-8化合物与式i-9化合物反应,得到式i-10化合物。该反应可在碱存在下进行。所述碱可选自叔丁醇钾、碳酸钾、碳酸钠、碳酸氢钠、碳酸铯、磷酸钾等。反应温度可以由本领域技术人员适当设定,例如可为25~120℃。
[0057]
步骤(g)中,将式i-10化合物脱除p3保护基,得到式i-11化合物。反应条件可采用本领域常用的脱除羟基保护基的反应条件。
[0058]
步骤(h)中,将式i-11化合物与式i-12化合物反应,得到式ia化合物。反应可在缩合剂的存在下进行。缩合剂可以是本领域常用的酸胺缩合剂,例如1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐、n,n-二异丙基乙胺、2-(7-氧化苯并三氮唑)-n,n,n’,n
’-
四甲基脲六氟膦酸酯等。反应温度可以由本领域技术人员适当设定,例如可为0~100℃。
[0059]
本发明一些实施方式中,n=1,通式(i)化合物可用通式(ib)表示,其制备可采用如下通用合成路线2:通用合成路线2
其中,r1、r2、r3、r4的定义如上所述。
[0060]
步骤(m)中,将式i-8化合物与氯乙酸乙酯反应,得到式i-12化合物。反应溶剂可为乙腈、丙酮、四氢呋喃、二氧六环、n,n-二甲基甲酰胺等。反应可在碱存在下进行。所述碱可选自:碳酸铯、碳酸钾、碳酸钠、氢氧化钾、氢氧化钠等。反应温度可以由本领域技术人员适当设定,例如可为25~150℃。
[0061]
步骤(n)中,将式i-12化合物水解,得到式i-13化合物。反应溶剂可为乙腈、丙酮、四氢呋喃、二氧六环、n,n-二甲基甲酰胺等与水的组合。反应可在碱存在下进行。所述碱可选自:氢氧化锂、氢氧化钠、氢氧化钾等。反应温度可以由本领域技术人员适当设定,例如可为25~120℃。
[0062]
步骤(o)中,将式i-13化合物与式i-11化合物反应,得到式ib化合物。反应可在缩合剂的存在下进行。缩合剂可以是本领域常用的酸胺缩合剂,例如1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐和n,n-二异丙基乙胺等。反应温度可以由本领域技术人员适当设定,例如可为0~100℃。
[0063]
另外,也可以直接使式i-12化合物酰胺化,得到式ib化合物。
[0064]
通式(i)化合物的应用
[0065]
通式(i)化合物可作为胃酸分泌抑制剂。
[0066]
通式(i)化合物可作为h
+
/k
+-腺苷三磷酸酶(h
+
/k
+-atpase)抑制剂。
[0067]
通式(i)化合物可作为钾离子竞争性酸阻滞剂(p-cabs)。
[0068]
通式(i)化合物可用于治疗和/或预防消化性溃疡、卓-艾综合征、胃炎、糜烂性食管炎、反流性食管炎、症状性胃食管反流疾病、巴雷特食管炎、功能性消化不良、幽门螺旋杆菌感染、胃癌、胃malt淋巴瘤、非甾体抗炎药引起的溃疡或手术后应激导致的胃酸过多或溃疡;或者抑制消化性溃疡、急性应激性溃疡、出血性胃炎或侵入性应激造成的上消化道出血。上述消化性溃疡包括但不限于胃溃疡、十二指肠溃疡或吻合口溃疡。症状性胃食管反流疾病包括但不限于非糜烂性的反流性疾病或无食管炎的胃食管反流疾病。
[0069]
药物组合物
[0070]
本发明的药物组合物包含有效量的通式(i)所示的化合物或其互变异构体、对映异构体、非对映异构体、及其混合物形式、及其可药用的盐、及其药学上可接受的载体或赋形剂或稀释剂。
[0071]“有效量”意指本发明化合物:(i)治疗特定疾病、病症或障碍,(ii)减弱、改善或消除特定疾病、病症或障碍的一或多种症状,或(iii)预防或延迟本文所述特定疾病、病症或障碍的一或多种症状发作的量。
[0072]
药学上可以接受的载体部分例子有纤维素及其衍生物(如羧甲基纤维素钠、乙基纤维素钠、纤维素乙酸酯等)、明胶、滑石、固体润滑剂(如硬脂酸、硬脂酸镁)、硫酸钙、植物油(如豆油、芝麻油、花生油、橄榄油等)、多元醇(如丙二醇、甘油、甘露醇、山梨醇等)、乳化剂(如)、润湿剂(如十二烷基硫酸钠)、着色剂、调味剂、稳定剂、抗氧化剂、防腐剂、无热原水等。
[0073]
本发明化合物或药物组合物的施用方式没有特别限制,代表性的施用方式包括(但并不限于):口服、瘤内、直肠、肠胃外(静脉内、肌肉内或皮下)、和局部给药。
[0074]
本发明化合物可以单独给药,或者与其他药学上可接受的化合物联合给药。
[0075]
本发明的另一方面涉及一种抑制胃酸分泌的方法,该方法包括给予需要治疗的患者有效剂量的通式(i)所示的化合物或其互变异构体、对映异构体、非对映异构体、及其混合物形式、及其可药用的盐或其药物组合物。
[0076]
本发明的另一方面涉及一种抑制h
+
/k
+-腺苷三磷酸酶(h
+
/k
+-atpase)的方法,该方法包括给予需要治疗的患者有效剂量的通式(i)所示的化合物或其互变异构体、对映异构体、非对映异构体、及其混合物形式、及其可药用的盐或其药物组合物。
[0077]
下面进一步例举实施例以详细说明本发明。同样应理解,以下实施例只用于对本发明进行进一步说明,不能理解为对本发明保护范围的限制,本领域的技术人员根据本发明的上述内容作出的一些非本质的改进和调整均属于本发明的保护范围。下述示例具体的工艺参数等也仅是合适范围中的一个示例,即本领域技术人员可以通过本文的说明做合适的范围内选择,而并非要限定于下文示例的具体数值。
[0078]
化合物的结构是通过核磁共振(nmr)或质谱(ms)来确定的,化合物的纯度是通过液相高压色谱仪(hplc)测定的。nmr的测定是用bruker avance-400核磁共振仪,溶剂为氘代二甲基亚砜(dmso-d6)或氘代甲醇(meoh-d4),内标为四甲基硅烷(tms),化学位移以ppm为单位。ms的测定使用安捷伦6120质谱仪。hplc使用安捷伦1200dad高压液相色谱仪测定。
[0079]
实施例1:n-环丙基-3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)丙酰胺
[0080]
第一步:2-(间甲苯基氧基)四氢-2h-吡喃在500ml三口瓶中依次加入间甲酚1a(104g,0.96mol)、对甲苯磺酸吡啶盐(24.1g,0.096mol)及二氯甲烷(250ml)。换气(氮气)三次后,氮气保护下,小心加入3,4-二氢-2h-吡喃(97.2g,1.15mol),室温搅拌2小时。tlc显示反应已完成,向反应液中加入水(80ml),萃取分离。有机相依次用水(40ml)及盐水(40ml)洗,然后用无水硫酸钠干燥、过滤及浓缩。粗品用硅胶柱(石油醚/乙酸乙酯=20/1)得无色油状物2-(间甲苯基氧基)四氢-2h-吡喃1b(141.1g),产率:76.6%。1h nmr(400mhz,cdcl3)δ
7.15(t,j=8.0hz,1h),6.87-6.84(m,2h),6.78(d,j=8.0hz,1h),5.39(s,1h),3.94-3.88(m,1h),3.61-3.56(m,1h),2.31(s,3h),2.03-1.98(m,1h),1.84-1.81(m,2h),1.65-1.60(m,3h)。
[0081]
第二步:2-(3-(溴甲基)苯氧基)四氢-2h-吡喃在500ml茄形瓶中依次加入2-(间甲苯基氧基)四氢-2h-吡喃1b(32g,166.45mmol)、四氯化碳(200ml)及n-溴代丁二酰亚胺(32g,179.77mmol),氮气置换三次。氮气保护下,小心加入偶氮二异丁腈(31.5g,192mmol),加热至100℃回流搅拌3个小时。tlc显示反应完成,将反应恢复至室温,过滤除去不溶物;滤液再静置2小时,再次过滤,滤液蒸干得到2-(3-(溴甲基)苯氧基)四氢-2h-吡喃1c(42g,淡黄色油状物),产率:77.8%。1hnmr(400mhz,cdcl3)δ7.18(t,j=8.0hz,1h),7.02(s,1h),6.93(d,j=8.0hz,1h),5.36(s,1h),4.39(s,2h),3.81(t,j=8.0hz,1h),3.51(t,j=8.0hz,1h),1.97-1.91(m,1h),1.80-1.78(m,2h),1.65-1.61(m,4h)。
[0082]
第三步:3-碘-2,6-二氢吡咯并[3,4-c]吡唑-5(4h)-羧酸叔丁酯在装有机械搅拌及冷凝装置的500ml三口瓶中依次加入2,6-二氢吡咯并[3,4-c]吡唑-5(4h)-羧酸叔丁酯1d(40g,191mmol)、1,2-二氯乙烷(300ml)及n-碘代丁二酰亚胺(64.6g,287mmol),氮气置换三次。油浴加热至80℃回流搅拌过夜。lcms及tlc均显示反应已完成,将反应恢复至室温,过滤除去不溶物,滤液蒸干。所得粗品经combi-flash(正相硅胶柱,80g,石油醚/二氯甲烷=0-40%)纯化得3-碘-2,6-二氢吡咯并[3,4-c]吡唑-5(4h)-羧酸叔丁酯1e(45g,黄色固体),产率:70.3%。ms m/z(esi):336.0[m+h]。
[0083]
第四步:3-碘-2-(3-((四氢-2h-吡喃-2-基)氧基)苄基)-2,6-二氢吡咯并[3,4-c]吡唑-5(4h)-羧酸叔丁酯向100ml单口瓶中依次放入3-碘-2,6-二氢吡咯并[3,4-c]吡唑-5(4h)-羧酸叔丁酯1e(4.65g,13.88mmol)、2-(3-(溴甲基)苯氧基)四氢-2h-吡喃1c(4.52g,16.66mmol)、碳酸铯(9.0g,27.76mmol)及乙腈(40ml),氮气换气3次,置于80℃油浴中反应3小时。反应结束后,将反应液过滤,滤液浓缩。残留物用combi-flash(正相硅胶柱,25g,石油醚/乙酸乙酯=0-30%)纯化,得到3-碘-2-(3-((四氢-2h-吡喃-2-基)氧基)苄基)-2,6-二氢吡咯并[3,4-c]吡唑-5(4h)-羧酸叔丁酯1f(2.55g,淡黄色固体,产率35.0%)。ms m/z(esi):526.0[m+h]。
[0084]
第五步:3-(2-氟苯基)-2-(3-((四氢-2h-吡喃-2-基)氧基)苄基)-2,6-二氢吡咯并[3,4-c]吡唑-5(4h))-甲酸乙酯在100ml单口瓶中依次加入3-碘-2-(3-((四氢-2h-吡喃-2-基)氧基)苄基)-2,6-二氢吡咯并[3,4-c]吡唑-5(4h)-羧酸叔丁酯1f(2.34g,4.46mmol)、(2-氟苯基)硼酸(0.75g,5.35mmol)、碳酸钠(1.18g,11.15mmol)、乙二醇二甲醚(8.0ml)及去离子水(4.0ml)。换气(氮气)三次后,氮气保护下,小心加入四(三苯基膦)钯(0.26g,0.223mmol),加完后,油浴升温到100℃,保温搅拌过夜。lcms显示反应已完成,将反应液降至室温,减压脱溶,残留物再加入水(10ml)及乙酸乙酯(60ml),萃取分离。有机相依次用水(15ml)及盐水(15ml)洗,然后用无水硫酸钠干燥、过滤及浓缩。粗品用combi-flash(正相硅胶柱,25g,石油醚:石油醚/乙酸乙酯
(体积比v/v=3/1
=0-40%得副产物,增大极性至70%)纯化,得到3-(2-氟苯基)-2-(3-((四氢-2h-吡喃-2-基)氧基)苄基)-2,6-二氢吡咯并[3,4-c]吡唑-5(4h))-甲酸乙酯1g(470mg,淡黄色油状物,产率21.4%)。ms m/z(esi):494.2[m+h]。
[0085]
第六步:3-((3-(2-氟苯基)-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯酚将三氟乙酸(4ml)加入到3-(2-氟苯基)-2-(3-((四氢-2h-吡喃-2-基)氧基)苄基)-2,6-二
二异丙基乙胺(313mg,2.43mmol),保温搅拌1小时。撤去冰浴,向上述反应液中加入环丙胺(247mg,4.32mmol),室温搅拌过夜。反应结束后,向反应液中加水(20ml),然后用二氯甲烷(100mlx 2)萃取分离。有机相合并后依次用水(20ml x 2)及饱和食盐水(30ml x 2)洗涤,然后使用无水硫酸钠干燥、过滤及浓缩,残留物用combi-flash(正相硅胶柱,25g,二氯甲烷/甲醇=0-10%)纯化得454mg粗品,取100mg再经prep-hplc制备(乙腈/水(含0.05%氨水)梯度冲洗),纯化得到n-环丙基-3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)丙酰胺1(50mg,无色油状物),产率:33.2%。ms m/z(esi):435.1[m+h]。1h nmr(400mhz,meod)δ7.49(q,j=8.0hz,1h),7.32(t,j=8.0hz,1h),7.26(d,j=8.0hz,2h),7.11(t,j=8.0hz,1h),6.77(d,j=8.0,8.0hz,1h),6.53(d,j=8.0hz,1h),6.47(s,1h),5.24(s,2h),4.10(t,j=6.0hz,1h),3.96(s,2h),3.87(s,2h),2.71(s,3h),2.68
–
2.63(m,1h),2.53(t,j=6.0hz,1h),0.74-0.68(m,2h),0.49-0.45(m,2h)。
[0090]
实施例2:3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)丙酰胺
[0091]
依次在微波管中加入3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)丙酸1k(100mg,0.253mmol)、2-(7-氧化苯并三氮唑)-n,n,n’,n
’-
四甲基脲六氟膦酸酯(144mg,0.380mmol)、n,n-二甲基甲酰胺(3ml)和氨的甲醇溶液(7m,0.8ml,5.57mmol),常温搅拌24h。反应结束后,反应液倒入水(5ml)中,然后用乙酸乙酯(20ml x 3)萃取。有机相合并后用饱和食盐水(10ml x 3)洗涤,然后使用无水硫酸钠干燥、过滤及浓缩,残留物通过tlc制备板纯化(二氯甲烷/甲醇=10/1),得到化合物3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)丙酰胺2(分子量:394.45,3.10mg,黄色固体)。ms m/z(esi):395.4[m+h]。1h nmr(400mhz,meod)δ7.52-7.46(m,1h),7.35-7.30(m,1h),7.27-7.21(m,2h),7.11(t,j=8.0hz,1h),6.77(d,j=7.6hz,1h),6.53(d,j=7.6hz,1h),6.49(s,1h),5.25(s,2h),4.11(t,j=6.0hz,2h),4.00(s,2h),3.91(s,2h),2.74(s,3h),2.61(t,j=6.0hz,2h)。
[0092]
实施例3:3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-甲基丙酰胺
[0093]
依次在25ml茄型瓶中加入3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)丙酸叔丁酯1j(25mg,0.0506mmol),二氯亚砜(1ml),置于40℃油浴中搅拌1小时。反应液浓缩,冰浴下加入甲胺溶液(27mg,0.870mmol)和四氢呋喃溶液(1ml),常温搅拌1小时。反应结束后,反应液浓缩,残留物通过tlc制备板纯化,得到化合物3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-甲基丙酰胺3(分子量:408.48,2.80mg,黄色固体),产率:10.8%。ms m/z(esi):409.4[m+h]。1h nmr(400mhz,meod)δ7.50-7.47(m,1h),7.36-7.29(m,1h),7.27-7.22(m,2h),7.11(t,j=8.0hz,1h),6.76(d,j=8.0hz,1h),6.52(d,j=7.2hz,1h),6.48(s,1h),5.24(s,2h),4.10(t,j=5.6hz,2h),3.93(s,2h),3.84(s,2h),2.73(s,3h),2.69(s,3h),2.57(t,j=5.6hz,2h)。
[0094]
实施例4:3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-(2-羟乙基)丙酰胺
[0095]
依次在微波管中加入3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)丙酸1k(100mg,0.253mmol)、2-(7-氧化苯并三氮唑)-n,n,n’,n
’-
四甲基脲六氟膦酸酯(144mg,0.380mmol)、n,n-二甲基甲酰胺(3ml)和乙醇胺(343mg,5.57mmol),常温搅拌24h。反应结束后,反应液倒入水(5ml)中,然后用乙酸乙酯(20ml x 3)萃取。有机相合并后用饱和食盐水(10ml x 3)洗涤,然后使用无水硫酸钠干燥、过滤及浓缩,残留物通过tlc制备板纯化(二氯甲烷/甲醇=10/1),得到化合物3-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-(2-羟乙基)丙酰胺4(31mg,黄色固体),产率:27.9%。ms m/z(esi):439.6[m+h]。1h nmr(400mhz,meod)δ7.47
(m,1h),7.30(m,1h),7.24(m,2h),7.09(m,1h),6.78(m,1h),6.53(m,2h),5.23(s,2h),4.12(t,2h),3.84(m,2h),3.75(m,2h),3.59(m,2h),3.30(m,2h),2.63(m,5h)。
[0096]
实施例5:2-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-甲基乙酰胺
[0097]
第一步:2-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)乙酸乙酯在25ml茄型瓶中依次放入3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯酚1i(1.41g,4.36mmol)、碳酸钾(1.81g,13.08mmol)及乙腈(10ml),搅拌30分钟后加入氯乙酸乙酯(1.10g,8.72mmol)。随后置于油浴中加热至70℃反应过夜。将反应液降至室温,倒入水(30ml)中,用乙酸乙酯(150ml x 2)萃取分离。有机相依次用水(20ml x 2)及饱和食盐水(20ml x 2)洗涤,经无水硫酸钠干燥、过滤及浓缩。残留物用combi-flash(正相硅胶柱,25g,二氯甲烷/甲醇=0-3%。增大极性至7%)纯化,得到2-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)乙酸乙酯5a(1.37g,黄色固体),产率76.8%。ms m/z(esi):410.2[m+h]。
[0098]
第二步:2-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)乙酸在25ml茄型瓶中依次加入2-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)乙酸乙酯5a(1.37g,3.35mmol)、氢氧化锂(400mg,16.75mmol)四氢呋喃(10ml)和水(6ml)。室温搅拌过夜后加入20ml水,用10ml二氯甲烷洗涤后,水相用1m稀盐酸中和至中性。用用乙酸乙酯(150ml x 2)萃取分离。有机相依次用水(20ml x 2)及饱和食盐水(20ml x 2)洗涤,经无水硫酸钠干燥、过滤及浓缩得到2-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)乙酸5b(1.08g,黄色固体),产率85.0%。ms m/z(esi):380.2[m-h]。
[0099]
第三步:2-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-甲基乙酰胺冰浴下,向25ml茄形瓶中依次加入2-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)乙酸5b(110mg,0.288mmol)、二氯甲烷(8.0ml)、1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐(127mg,0.334mmol)及n,n-二异丙基乙胺(313mg,2.43mmol),保温搅拌1小时。撤去冰浴,向上述反应液中加入甲胺盐酸盐(291mg,4.32mmol),室温搅拌过夜。反应结束后,向反应液中加水(20ml),然后用二氯甲烷(100ml x 2)萃取分离。有机相合并后依次用水(20ml x 2)及饱和食盐水(30ml x 2)
苯并三唑-1-基氧三吡咯烷基六氟磷酸盐(127mg,0.334mmol)及n,n-二异丙基乙胺(313mg,2.43mmol),保温搅拌1小时。撤去冰浴,向上述反应液中加入环丙胺(247mg,4.32mmol),室温搅拌过夜。反应结束后,向反应液中加水(20ml),然后用二氯甲烷(100ml x 2)萃取分离。有机相合并后依次用水(20ml x 2)及饱和食盐水(30ml x 2)洗涤,然后使用无水硫酸钠干燥、过滤及浓缩,残留物用combi-flash(正相硅胶柱,25g,二氯甲烷/甲醇=0-10%)纯化得到n-环丙基-2-(3-((3-(2-氟苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)乙酰胺7(50mg,无色油状物),产率:8.2%。ms m/z(esi):421.1[m+h]。1h nmr(400mhz,meod)δ7.49(m,1h),7.40(m,1h),7.27(m,1h),7.17(m,2h),6.92(m,1h),6.88(m,1h),6.73(m,1h),5.48(s,2h),4.45(2,2h),3.84(m,2h),2.97(m,1h),2.17(s,3h),0.86(m,2h),0.60(m,2h)。
[0104]
实施例8:3-(3-((3-(2-氯苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-(2-羟乙基)丙酰胺
[0105]
第一步:3-(3-((3-(2-氯苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-(2-羟乙基)丙酰胺
[0106]
依次在茄形瓶中加入2-(3-((四氢-2h-吡喃-2-基)氧基)苄基)-2,6-二氢吡咯并[3,4-c]吡唑-5(4h)-羧酸叔丁酯8a(1.0g,2.5mmol)、乙酸钾(1.47g,15mmol)、氯化烯丙基钯(ii)二聚物(91mg,0.25mmol)、n,n-二甲基乙酰胺(25ml)及1-氯-2-碘苯(1.2g,5.0mmol)。油泵换气(氩气)四次后放入提前升温到100℃的油浴中反应2小时。反应恢复至室温后,把反应液直接倒入水(40ml)中,用乙酸乙酯(100ml x 2)萃取。有机相用盐水(40ml x 2)洗,然后用无水硫酸钠干燥、过滤及浓缩。粗品经combi-flash(正相硅胶柱,80g,石油醚:石油醚/乙酸乙酯
(体积比v/v=10/1
=0-100%至石油醚:石油醚/乙酸乙酯
(体积比v/v=5/1
=100%)分离得到3-(3-((3-(2-氯苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯
氧基)-n-(2-羟乙基)丙酰胺8b(187mg,黄色油状物),产率:14.7%。ms m/z(esi):510.3[m+h]。1h nmr(400mhz,cdcl3)δ7.48(d,j=8.0hz,1h),7.36(t,j=8.0hz,1h),7.31
–
7.25(m,1h),7.21-7.19(m,1h),7.13(t,j=8.0hz,1h),6.91(d,j=8.0hz,1h),6.67(d,j=8.0hz,1h),6.58(d,j=8.0hz,1h),5.30(t,j=4.0hz,1h),5.13(s,2h),4.58(s,1h),4.51(s,1h),4.41(s,1h),4.37(s,1h),3.84(t,j=10.0hz,1h),3.58-3.53(m,1h),2.02
–
1.90(m,2h),1.88
–
1.76(m,2h),1.49(s,9h)。
[0107]
第二步:3-((3-(2-氯苯基)-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯酚
[0108]
将三氟乙酸(0.5ml)加入到3-(3-((3-(2-氯苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-(2-羟乙基)丙酰胺8b(187mg,0.367mmol)的二氯甲烷(1.0ml)溶液中,室温反应1小时。反应结束后,直接浓缩得到粗品3-((3-(2-氯苯基)-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯酚8c(100mg,棕色油状物),产率:94%。ms m/z(esi):326.1[m+h]。
[0109]
第三步:3-((3-(2-氯苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯酚将甲醛水溶液(37%w/w,70mg,2.2mmol)加入到3-((3-(2-氯苯基)-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯酚8c(100mg,0.30mmol)的甲醇(3ml)溶液中,室温搅拌半小时。将冰醋酸(0.5ml)及醋酸硼氢化钠(311mg,1.47mmol)缓慢加入反应液中,再室温反应过夜。反应液浓缩后用二氯甲烷(90ml)溶解稀释,再依次用氨水/水(10ml x 2,1/5)、饱和食盐水(20ml)洗涤,有机相用无水硫酸钠干燥、浓缩。残留物用combi-flash(正相硅胶柱,20g,二氯甲烷:二氯甲烷/甲醇混合溶剂
(体积比v/v=10/1
)=0-50%)分离得到3-((3-(2-氯苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯酚8d(165mg,黄色油状物),产率:90.0%。ms m/z(esi):340.1[m+h]。
[0110]
第四步:3-(3-((3-(2-氯苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)丙酸取10ml单口瓶,依次加入3-((3-(2-氯苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯酚8d(78mg,0.23mmol)及n,n-二甲基甲酰胺(2ml),开动搅拌,n2置换三次。n2保护下小心加入氢化钠(18mg,0.46mmol),加完后,继续室温搅拌半小时,再缓慢加入溶于n,n-二甲基甲酰胺(4ml)的6(42mg,0.28mmol),反应结束后,加水(2ml)淬灭,然后在冰浴下用1n稀盐酸调ph=4-5,再用二氯甲烷(20ml x2)萃取。有机相合并后用饱和食盐水(6ml x 3)洗涤,然后使用无水硫酸钠干燥、过滤及浓缩,得到化合物3-(3-((3-(2-氯苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)丙酸8e(100mg,淡黄色固体),产率:90%。ms m/z(esi):412.2[m+h]。
[0111]
第五步:3-(3-((3-(2-氯苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-(2-羟乙基)丙酰胺在10ml茄型瓶中依次加入3-(3-((3-(2-氯苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)丙酸8e(70mg,0.17mmol),n2置换三次。n2保护下小心加入氯化亚砜(1ml),室温搅拌5分钟,再用1ml注射器滴入1小滴n,n-二甲基甲酰胺,缓慢升温至80℃,并保温搅拌1.5小时。减压除去多余氯化亚砜,并用干燥二氯甲烷(3ml x 2)带2次。残留物溶于干燥二氯甲烷(3ml),小心滴加至放有乙醇胺(52mg,0.34mmol)、三乙胺(69mg,0.68mmol)及干燥二氯甲烷(3ml)的10ml反应瓶中(冰浴冷却及n2保护),滴完后,继续0℃搅拌15分钟。撤去冰浴,室温搅拌2小时,lcms显示反应已完成。加水(10ml)淬灭,用二氯甲烷(20ml x 2)萃取。有机相合并后用饱和食盐水(6ml x 3)洗涤,然
后使用无水硫酸钠干燥、过滤及浓缩,所得粗品经prep-hplc制备(乙腈/水(含0.05%氨水)梯度冲洗),纯化得到3-(3-((3-(2-氯苯基)-5-甲基-5,6-二氢吡咯并[3,4-c]吡唑-2(4h)-基)甲基)苯氧基)-n-(2-羟乙基)丙酰胺8(1.21mg,黄色固体,产率15.1%)。ms m/z(esi):455.4[m+h]。1h nmr(400mhz,meod)δ7.53(d,j=8.0hz,1h),7.44(t,j=8.0hz,1h),7.36(t,j=8.0hz,1h),7.30(d,j=8.0hz,1h),7.10(t,j=8.0hz,1h),6.76(d,j=8.0hz,1h),6.49(d,j=8.0hz,1h),6.44(s,1h),5.34(t,j=4.0hz,1h),5.14(s,2h),4.58(s,3h),4.11(t,j=6.0hz,1h),3.84(s,1h),3.72(s,1h),3.59(t,j=6.0hz,1h),3.34(s,h),3.13(s,1h),2.62(s,3h)。
[0112]
测试例:化合物对h
+
/k
+
atpase酶活性抑制的测定下面的实验是用来测定本发明化合物对h
+
/k
+
atpase酶活性的抑制作用。
[0113]
1.实验材料plate reader:spectramax m5(md)孔雀石绿(sigma aldrich,213020-25g)钼酸铵(sigma aldrich,277908-20g)atp(sigma aldrich,a1852-1vl)。
[0114]
2.缓冲液配制酶工作液:对酶进行滴定,用缓冲液1将酶稀释,反应时,取5μl稀释液至50μl的反应体系中atp溶液:100mm的atp用无k
+
buffer稀释至5mm,取5μl稀释液至50μl的反应体系中,即atp的终浓度为500μmmlg显色液:将0.12%的mlg、7.5%钼酸铵、11%的tween-20按100:25:2的体积混匀,检测时每孔加入15μl缓冲液1:50mm tris-hcl ph 6.5,5mm氯化镁(magnesium chloride),10μm缬氨霉素(valinomycin)缓冲液2:50mm tris-hcl ph 6.5,5mm氯化镁(magnesium chloride),10μm缬氨霉素(valinomycin),20mm kcl匀浆缓冲液:10mmol/l tris-hcl,ph 6.8,0.25m蔗糖(sucrose),1mmol/l edta7.5%ficoll分层液:匀浆缓冲液+7.5%(w/w)400(聚蔗糖400)。
[0115]
3.实验步骤
[0116]
3.1.h
+
/k
+
atp酶提取
[0117]
(1)分离出兔胃组织,自来水冲洗血迹,食物残留。
[0118]
(2)利用预冷的nacl溶液彻底清洗胃底部位,去除表面粘液。
[0119]
(3)将剥离的粘膜,装于样品袋或50ml离心管,迅速冻于液氮罐中。
[0120]
(4)取出组织,用手术剪刀剪碎,加入预冷的匀浆缓冲液(4ml/g组织),于组织匀浆机中匀浆2-10min。
[0121]
(5)匀浆后,如果有较大的组织颗粒,可离心(600g,10min)去除,然后将上清移至干净的离心管中,20000g离心30min后,然后将上清移至干净的离心管中,进一步离心,100000g离心90min,收集沉淀。
[0122]
(6)利用匀浆缓冲液重悬沉淀,吹散均匀,等比例加入7.5%ficoll分层液,
100000g离心90min,收集沉淀。
[0123]
(7)匀浆缓冲液重悬沉淀,吹散均匀,用bradford测蛋白浓度。分管冻存于-80℃备用。
[0124]
3.2.h
+
/k
+
atp酶活性实验
[0125]
(1)每个实验孔中加入35μl反应缓冲液,再加入35μl的缓冲液1
[0126]
(2)全酶和缓冲液孔中,加入5μl含10%dmso的缓冲液1
[0127]
(3)化合物孔中,加入5μl 10x化合物工作液混匀
[0128]
(4)缓冲液孔中加入5μl的缓冲液1
[0129]
(5)其余孔中,加入5μl 10x酶工作液混匀于37℃孵育30min
[0130]
(6)向所有实验孔中加入5μl 10xatp工作液,并混匀于37℃孵育20min
[0131]
(7)向所有实验孔中加入15μl mlg显色液,并混匀于室温孵5-30min
[0132]
(8)m5仪器检测620nm的读数
[0133]
4.数据分析抑制率用以下公式计算:抑制率(ic
50
)=【od(样品孔)-od(含氯化钾的全酶孔)】/【(od(含氯化钾的全酶孔)-(od(不含氯化钾的全酶孔)】
×
100%
[0134]
5.实验结果
[0135]
各实施例化合物的抑制率(ic
50
)在表2中示出表2化合物编号ic
50
(μm)实施例10.08978实施例20.3889实施例30.1646实施例40.4672实施例50.3478实施例60.7468实施例70.5078实施例81.864
[0136]
从表2可以看出,本发明化合物具有优异的h
+
/k
+
atpase酶抑制活性,可用于制备胃酸分泌抑制剂。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1