一种小叶金钱草抗炎化合物及其制备方法与流程
本发明涉及抗炎化合物及其制备方法,具体地说是小叶金钱草抗炎化合物及其制备方法。
背景技术:
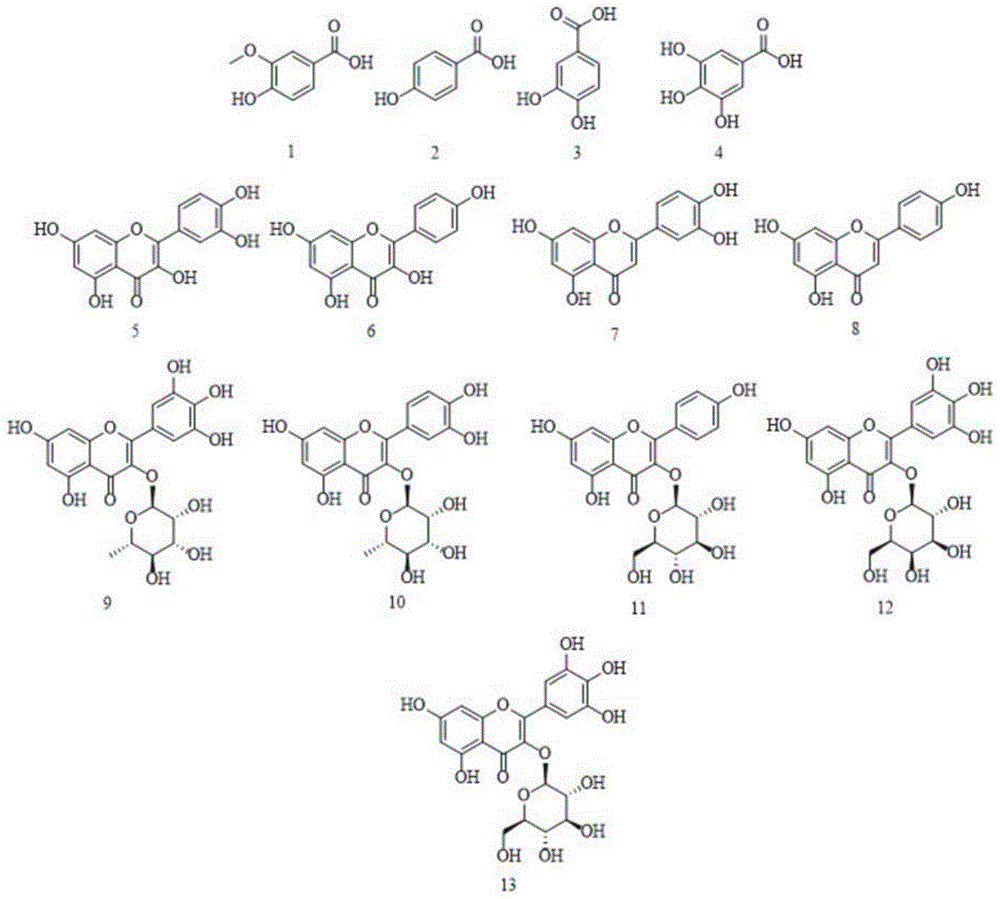
:小叶金钱草(hydrocotylesibthorpioideslam.),为伞形科天胡荽属植物天胡荽,中国传统中药之一。具备多种清热解毒、跌打淤肿以及消肿等功效,民间常用于治疗黄疸型肝炎,临床上常用于治疗结石、黄疸性肝炎及胆囊炎等症。前期研究表明药材中富含丰富的黄酮类化合物。同时药理实验也证实抗氧化、抗病毒以及抑菌消炎等作用。但截至目前,小叶金钱草只作为民间用药,尚未被中国药典收录,因此对小叶金钱草化学成分及药理活性的深入研究具有重要意义。小叶金钱草通常生长在海拔475-3000米湿润的草地、河沟边和林下,广泛分布于中国的许多地区,如:产于陕西、江苏、安徽、浙江、江西、福建、湖南、湖北、广东、广西、台湾,四川、贵州、云南等省区,通常生长在湿润的草地、河沟边、林下,朝鲜、日本、东南亚至印度也有分布,本种为本属分布最广的一种,可全草入药,具有清热解毒、保肝利胆、治黄疸、痢疾、目翳、喉肿和跌打损伤等功效。多年生草本植物,有气味。茎细长而匍匐,平铺地上成片,节上生根,叶片膜质至草质,圆形或肾圆形,长0.5-1.5厘米,宽0.8-2.5厘米,基部心形,两耳有时相接,不分裂或5-7裂,裂片阔倒卵形,边缘有钝齿,表面光滑,背面脉上疏被粗伏毛,有时两面光滑或密被柔毛,叶柄长0.7-9厘米,无毛或顶端有毛,托叶略呈半圆形,薄膜质,全缘或稍有浅裂。伞形花序与叶对生,单生于节上,花序梗纤细,长0.5-3.5厘米,短于叶柄1-3.5倍,小总苞片卵形至卵状披针形,长1-1.5毫米,膜质,有黄色透明腺点,背部有1条不明显的脉,小伞形花序有花5-18,花无柄或有极短的柄,花瓣卵形,长约1.2毫米,绿白色,有腺点,花丝与花瓣同长或稍超出,花药卵形,花柱长0.6-1毫米,果实略呈心形,长1-1.4毫米,宽1.2-2毫米,两侧扁压,中棱在果熟时极为隆起,幼时表面草黄色,成熟时有紫色斑点,花果期4-9月。现有技术存在的问题:目前小叶金钱草成分分离方面主要通过柱层析手段,但所需分离时间长、损耗溶剂多以及样品不可逆吸附等。因此建立快速有效的分离方法意义重大。研究以高效液相色谱为基础,对乙酸乙酯萃取物指纹图谱进行研究,并通过lc-ms/ms对其复杂化学成分进行初步鉴定。其次,通过层析分离(葡聚糖凝胶、反相硅胶和大孔树脂等)与高速逆流色谱相结合的方法,对乙酸乙酯和正丁醇萃取物化学成分进行分离,成功从小叶金钱草中分离出包含同分异构体在内的黄酮苷、酚类等化合物,并通过核磁共振波谱对有机化合物特征信号进行总结研究,为分离的天然有机化合物提供新思路以及对微量成分的快速富集提供借鉴。技术实现要素:鉴于现有技术的不足,本发明提供了小叶金钱草抗炎化合物及其制备方法。为了达到上述目的,本发明采用了如下的技术方案:小叶金钱草抗炎单体化合物,包括:香草酸(1)、4-羟基苯甲酸(2)、原儿茶酸(3)、没食子酸(4)、槲皮素(5)、山柰酚(6)、木犀草素(7)、芹菜素(8)、杨梅苷(9)、槲皮苷(10)、紫云英苷(11)、杨梅素3-o-半乳糖苷(12)、异杨梅树皮苷(杨梅素3-o-葡萄糖苷)(13)和槲皮素3,3'-二-α-l-鼠李糖苷(16),其中抗炎活性最强的为4-羟基苯甲酸(2)。其中,4-羟基苯甲酸(2)、原儿茶酸(3)、杨梅苷(9)、槲皮素3,3'-二-α-l-鼠李糖苷(16)能够显著抑制白细胞介素(il-6)的增加;香草酸(1)、4-羟基苯甲酸(2)、木犀草素(7)能够显著抑制raw264.7巨噬细胞肿瘤坏死因子(tnf-α)的增加,具有显著抗炎作用。附图说明图1:化合物1-13的结构图;图2:化合物14-17的结构图;图3:小叶金钱草乙酸乙酯萃取物hplc-esi-ms正离子模式下质谱信息,信号强度及初步鉴定结果;图4:小叶金钱草乙酸乙酯萃取物hplc-esi-ms负离子模式下质谱信息,信号强度及初步鉴定结果;图5:elisa试剂盒tnf-α(a)、il-6(b)标准品四参数logistic拟合曲线图。具体实施方式为使发明的目的、技术方案和优点更加清楚,下面结合附图对本发明的具体实施方式进行详细说明。这些优选实施方式的示例在附图中进行了例示。附图中所示和根据附图描述的本发明的实施方式仅仅是示例性的,并且本发明并不限于这些实施方式。在此,还需要说明的是,为了避免因不必要的细节而模糊了本发明的技术方案,在附图中仅仅示出了与根据本发明的方案密切相关的结构和/或处理步骤,而省略了关系不大的其他细节。实施例1如附图1-2所示,该实施例提供小叶金钱草抗炎单体化合物,包括:香草酸(1)、4-羟基苯甲酸(2)、原儿茶酸(3)、没食子酸(4)、槲皮素(5)、山柰酚(6)、木犀草素(7)、芹菜素(8)、杨梅苷(9)、槲皮苷(10)、紫云英苷(11)、杨梅素3-o-半乳糖苷(12)、异杨梅树皮苷(杨梅素3-o-葡萄糖苷)(13)和槲皮素3,3'-二-α-l-鼠李糖苷(16),其中抗炎活性最强的为4-羟基苯甲酸(2)。实施例2该实施例提供小叶金钱草乙酸乙酯萃取物hplc-esi-ms/ms分析方法,包括:2.1仪器与试剂安捷伦1100系列液相色谱仪(agilent公司,美国),配备在线脱气机,四元泵,高性能自动进样器,二极管阵列检测器,电喷雾电离源,离子阱质谱。2.2样品提取药材粉碎,过40目筛。取已粉碎药材5g于250ml锥形瓶中,置于kj-11030al型超声波清洗器,按液料比10:1加入70%(v:v)乙醇溶液,于600w、40khz、60℃提取1h,过滤除去药渣,将药渣重复提取4次。提取液经减压浓缩,得棕褐色黏稠稠物,即醇提浸膏。浸膏浸入蒸馏水超声辅助溶解,依次用10倍蒸馏水体积的石油醚、乙酸乙酯、正丁醇萃取各5次。浓缩乙酸乙酯萃取液,得小叶金钱草乙酸乙酯萃取物,4℃避光保存备用。取一定量醇提浸膏和乙酸乙酯萃取物,乙腈为溶剂超声溶解,0.45μm针头过滤器过滤后,进行hplc分析,其中产物的纯度按面积归一化法计算。2.3色谱质谱条件采用esi源,毛细管电压3500v,雾化气压力30psi,干燥气流速9l/min,干燥气温度350℃,毛细管电压3500v,质量数扫描范围100~1000。正离子模式下:液相色谱条件见表2.1,向流动相a中添加10mm甲酸-氨水缓冲液(调至偏酸性ph4.5)。负离子模式下:液相色谱条件表2.1,向流动相a添加10mm甲酸-氨水缓冲液(调至偏碱性ph7.5)。表2.1hplc-esi-ms/ms梯度洗脱程序时间(min)流动相a:乙腈(%)流动相b:水-0.1%甲酸(%)流速(ml/min)030701.004070301.00609551.002.4hplc-esi-ms/ms正负离子模式离子流色谱图采用色谱-质谱分析方法获得小叶金钱草乙酸乙酯萃取物正离子模式离子流色谱图相关信息,见表2.3(图3)。正离子模式下质谱分子离子峰强度大于1*106的质谱信号共有14个,负离子模式下质谱分子离子峰强度大于1*106的质谱信号共有14个(图4)。以正离子模式为例进行分析,通过质谱数据库以及文献信息查阅对可能对应的化合物进行初步鉴定,负离子模式鉴定结果与正离子模式类似。2.5小叶金钱草乙酸乙酯萃取物化学成分lc-ms/ms鉴定峰1:[m+h]+m/z123.6,未发现准分子离子碰撞离子质谱信号,其分子离子峰信号强度小于1*104,属微量成分,查阅质谱数据库可知,其元素组成可能为c6h5o2n,对应分子量为123.6的烟酸。峰2:[m+h]+m/z171.4,二级质谱未检出质谱碎片,其分子离子峰信号强度小于1*104,属微量成分,查阅质谱数据库可知,其元素组成可能为c7h6o5,对应分子量为170.0的五倍子酸,即没食子酸。峰3:[m+h]+质核比为139.5。查阅质谱数据库确定其元素组成为c7h6o3,分子量为138.3的有机酸类化合物或醌类化合物,对应化合物可能为:水杨酸、4-羟基苯甲酸、2-羟基-5-甲基对苯醌。峰4:[m+h]+质核比为155.5。查阅质谱数据库其元素组成可能为c8h10o3、c7h6o4,分子量为154.4的有机酸类化合物,对应化合物可能为:绵马次酸、原儿茶酸。峰5:[m+h]+m/z163.4,[2m+h+h]2+m/z326.5,[m+h+na]2+m/z348.7,查阅质谱数据库其元素组成可能为c9h6o3、c11h14o、c10h10o2,分子量为162.5数据库中涉及化合物较多,包括伞形内酯、2,2-二甲基色烷、黄樟脑,对应化合物类别为:香豆精类、色烷类以及其他天然有机化合物。峰6、峰8:[m+h]+m/z481.4,ms/ms318.5/319.4。查阅质谱数据库可知,为六羟基黄酮,母核化合物元素组成为c15h10o8。根据二级质谱失去162的质谱碎片,判定为糖残基碎片,对应糖苷分子量应为180,即葡萄糖和半乳糖,二者为同分异构体。因为仅根据质谱信息无法判断化合物具体结构,仅能判定化合物为黄酮苷类化合物,为单糖苷取代黄酮。峰7:[m+h]+质核比为169.4,其分子离子峰信号强度小于1*105,属微量成分。查阅质谱数据库其元素组成可能为c8h8o4、c10h12o3,分子量为168.3的有机酸类化合物或醌类化合物,对应化合物可能为:香草酸、3-羟基-2-异丙基-5-甲基对苯醌。峰9:[m+h]+m/z611.3,ms/ms178.7/358.0/477.1/478.0。通过查阅质谱数据库中m/z358.0对应的黄酮类化合物,列举如下:3′,4′,5,7-o-四甲基槲皮素、3,3′,4′,7-o-四甲基槲皮素和色曼酮,母核元素组成为c19h18o7,前两者为简单取代黄酮而色曼酮为鱼藤酮类化合物。根据二级质谱丢失碎片610-478=132,478-358=120,判定所失去碎片为糖残基碎片,为单糖失去一分子水形成。对应单糖为五碳醛糖(核糖、木糖、阿拉伯糖,m/z150.0)和脱氧核糖(m/z138.0)。但核糖与脱氧核糖在取代糖苷中并不出现,故化合物结构有待进一步分离研究。峰10:[m+h]+m/z611.3,ms/ms318.4/319.4/464.5/465.4。查阅质谱数据库可知为六羟基黄酮,母核化合物元素组成为c15h10o8。根据二级质谱失去碎片610-464=146,464-318=146的质谱碎片,判定为糖残基碎片,对应糖苷分子量应为164,即化合物带两分子鼠李糖。经文献查阅[37],化合物可能为杨梅素3,3'-二-α-l-鼠李糖苷,结论是否成立有待进一步分离验证。峰11:[m+h]+m/z319.6,ms/ms301.5。查阅质谱数据库其元素组成为c15h10o8,分子量为318的黄酮类化合物。通过查阅质谱数据库中m/z318对应的黄酮类化合物,对应的化合物可能为:3,3′,4′,5′,5,7-六羟基黄酮(即杨梅素)和3,3′,4′,5,6,7-六羟基黄酮。峰12:[m+h]+m/z465.3,ms/ms319.6。换句话说,峰12为峰11化合物的去糖苷化合物,即3,3′,4′,5′,5,7-六羟基黄酮(即杨梅素)和3,3′,4′,5,6,7-六羟基黄酮。根据二级质谱失去碎片465-319=146的质谱碎片,判定为糖残基碎片,对应糖苷分子量应为164,即化合物与一分子鼠李糖成苷。因为仅根据质谱信息无法判断化合物具体结构,仅能判定化合物为黄酮苷类化合物,为单糖苷取代黄酮。峰13:[m+h]+m/z595.4,ms/ms286.5/287.6/448/449.3。查阅质谱数据库可知为四羟基黄酮,母核化合物元素组成为c15h10o6。根据二级质谱失去碎片594-448=146,448-286=162的质谱碎片,判定为糖残基碎片,对应糖苷分子量应为164和180,即化合物带一分子鼠李糖和一分子葡萄糖/半乳糖。通过质谱无法判断化合物带一份子二糖还是两分子单糖。在已知文献中尚未发现相关报道,故化合物结构需要进一步鉴定研究。峰14:[m+h]+m/z449.5,ms/ms286.5。查阅质谱数据库m/z286化合物可知母核化合物为四羟基黄酮。根据二级质谱失去碎片448-286=162的质谱碎片,判定为糖残基碎片,对应糖苷分子量应为180,即化合物与一分子葡萄糖或半乳糖成苷。因为仅根据质谱信息无法判断化合物具体结构,仅能判定化合物为黄酮苷类化合物,为单糖苷取代黄酮。峰15:[m+h]+m/z595.4,ms/ms303.3/449.6。查阅质谱数据库可知为五羟基黄酮,母核化合物元素组成为c15h10o7。根据二级质谱失去碎片595-449=146,449-303=146的质谱碎片,判定为糖残基碎片,对应糖苷分子量应为164,即化合物带两分子鼠李糖。经文献查阅,化合物可能为槲皮素3,3'-二-α-l-鼠李糖苷,结论是否成立有待进一步分离验证。峰16:[m+h]+m/z449.5,ms/ms302.5。查阅质谱数据库m/z302化合物可知母核化合物为五羟基黄酮。根据二级质谱失去碎片448-302=146的质谱碎片,判定为糖残基碎片,对应糖苷分子量应为164,即化合物与鼠李糖成苷。因为仅根据质谱信息无法判断化合物具体结构,仅能判定化合物为黄酮苷类化合物,为单糖苷取代黄酮。峰17:[m+h]+m/z447.5,ms/ms270.6。查阅质谱数据库m/z270化合物可知母核化合物为三羟基黄酮。根据二级质谱失去碎片446-270=176的质谱碎片,判定为糖残基碎片,对应糖苷分子量应为194,即化合物与葡萄糖醛酸或半乳糖醛酸成苷。因为仅根据质谱信息无法判断葡萄糖醛酸或半乳糖醛酸连接位置,仅能判定化合物为黄酮苷类化合物,为单糖苷取代黄酮。峰18:[m+h]+m/z463.5,ms/ms286.3。查阅质谱数据库m/z286化合物可知母核化合物为四羟基黄酮。根据二级质谱失去碎片462-286=176的质谱碎片,判定为糖残基碎片,对应糖苷分子量应为194,即化合物与葡萄糖醛酸或半乳糖醛酸成苷。因为仅根据质谱信息无法判断葡萄糖醛酸或半乳糖醛酸连接位置,仅能判定化合物为黄酮苷类化合物,为单糖苷取代黄酮。峰19:[m+h]+m/z287.4,它们对应天然产物中的简单黄酮:木犀草素(c15h10o6)、山柰酚(c15h10o6)和去甲基杜鹃素(c16h14o5),山柰酚与木犀草素是c-3/c-3’连接羟基的同分异构体。峰20:[m+h]+m/z303.5,它们也同样对应天然产物中的简单黄酮:槲皮素(c15h10o7)、3,5,7-三甲基-4′-甲氧基黄烷酮(c16h14o6)、3′,7-二羟基-2′,4′-二甲氧基黄烷(c17h18o5)。峰21:[m+h]+m/z271.4,有两种组成形式的化合物:3,5,7-三羟基黄酮、黄芩黄酮、染料木素、芹菜素(c15h10o5)和球松素、2′,6′-二羟基-,4′-甲氧基查耳酮(c16h14o4)。仅根据质谱提供的信息无法鉴定化合物的具体结构,需要进一步分离鉴定。通过紫外光谱,esi-ms/ms正离子模式和负离子模式研究,对小叶金钱草次生代谢产物成分有了初步的了解,为进一步开发植物资源,科学评价药用及开发利用提供科学依据和技术支持。通过数据库比对可以鉴定出部分化合物,如简单黄酮类和结构较为简单的有机酸类化合物。而对于黄铜苷类成分,质谱则不能起到鉴定的作用,只能做出初步的推测以及判断。但质谱的初步鉴定将为下一步药材化学成分的分离奠定了一定的工作基础。实施例3该实施例提供小叶金钱草乙酸乙酯萃取物化学成分分离与结构鉴定,包括如下步骤:3.1样品提取与前处理re.52aa旋转蒸发器,上海亚荣生化仪器厂;真空脱气过滤装置,美国waters公司;kj-11030al型超声波清洗器,深圳市科洁超声科技有限公司;waters600高效液相色谱仪,美国waters公司;waters2998pad二极管阵列检测器,美国waters公司;hsccctbe-300b型高速逆流色谱仪(柱体积300ml),中国上海同田生化技术有限公司;n2000双通道色谱工作站,浙江大学智达信息工程有限公司;葡聚糖凝胶(sephadexlh-20),安徽博美生物科技有限公司;氯仿、正己烷、乙酸乙酯、正丁醇、乙醇、乙腈、甲酸,分析纯,高效液相色谱所用的乙腈为色谱纯,天津市百世化工有限公司;水,二次蒸馏水。实验考察了甲醇-水和乙腈-水作为流动相时对样品中化学成份的洗脱能力,发现乙腈-水系统更适用于样品中化学成分的分离分析。同时考查了乙腈-水不同梯度条件下的色谱分离结果,确定最终梯度洗脱条件。色谱柱:waters600sunfiretmc18(4.6mm×250mm,5μm),柱温为25℃,流速为1.0ml/min,紫外检测波长λ=254nm,同时使用pad在线扫描功能获得色谱峰紫外吸收光谱,紫外光谱扫描范围:200-400nm。进样10μl分析。表3.1小叶金钱草乙酸乙酯提取物梯度洗脱程序时间(min)流动相a:乙腈(%)流动相b:水-0.1%甲酸(%)流速(ml/min)015851.007040601.007540601.00将购置的药材经中药粉碎机粉碎后过40目筛。称取20kg粉末分次置于kj-11030al型超声波清洗器,按液料比10:1加入70%(v:v)乙醇溶液,于600w、40khz、60℃提取1h,过滤除去药渣,将药渣重复提取4次。提取液减压浓缩,得棕褐色黏稠浸膏。浸膏用蒸馏水混悬并超声溶解,10倍体积石油醚、乙酸乙酯、正丁醇萃取5次。浓缩乙酸乙酯萃取液,冻干得乙酸乙酯萃取物。4℃避光保存备用。取3g乙酸乙酯萃取物,用30ml蒸馏水超声溶解并过0.45μm滤膜,作为分离样品一,即f1。取5g乙酸乙酯萃取物,用30ml蒸馏水超声溶解并过0.45μm滤膜。经sephadexlh-20柱层析分离(3×80cm),分别用水、10%、20%、30%、40%、50%、75%、100%甲醇洗脱,每个比例洗脱2个柱体积,1/20柱体积为一馏分。收集30%,40%,75%和100%甲醇洗脱馏分,合并色谱峰保留时间相同的馏分。经hplc检测,30%洗脱液合并相同馏分后得化合物3,结构鉴定见3.6。40%脱液合并相同馏分后得化合物4和16,结构鉴定见3.6。75%脱液合并相同馏分,获得三组混合物,即三组乙酸乙酯分离样品f2-f4。100%洗脱液合并相同馏分后得化合物7,8以及两化合物的混合物,混合物经反向硅胶ods/rp-18(2×100cm)层析分离,分别用30%,50%、70%和100%甲醇梯度洗脱,得化合物5和6,结构鉴定见3.6。将分离样品f1-f4旋蒸除去溶剂并于-70℃冷冻干燥24h,得到hsccc分离样品。上述色谱条件下,醇提浸膏和乙酸乙酯萃取物(f1样品)的hplc色谱分离。从醇提浸膏和乙酸乙酯提取物色谱图中,选取显著的色谱峰进行深入研究。经过乙酸乙酯萃取后,部分化合物被明显富集,所包含化学成分较为复杂,主要成分有11个,其保留时间分别为:3.835min,5.860min,9.493min,10.802min,15.511min,19.009min,,19.775min,20.567min,24.721min,26.183min,31.668min,其余成分为此要及微量成分,各吸收峰的紫外光谱图。根据hplc-pad紫外光谱初步分析可知,保留时间3.830min,5.870min,9.627min和10.752min色谱峰的λmax在254nm,260nm和270nm,符合共轭体系苯环衍生物即有机酸类化合物的紫外光谱吸收特征,其余成分以及其他化学成分的λmax在在峰带i:300-400nm及峰带ii:220-280nm有明显紫外吸收,故所属化合物类别为黄酮类化合物。上述的色谱条件下,初步将主色谱峰定为分离目标,其中f2分离样品主要色谱峰有两个,其保留时间分别为:18.974min和26.282min。f3分离样品主要色谱峰有两个,其保留时间分别为:19.958min和20.239min。f4分离样品中主色谱峰有三个,其保留时间分别为20.320min、23.593min和25.746min。与乙酸乙酯萃取物相比,经sehpadexlh-20纯化后,大部分杂质以及其他化合物得到较好的初步分离,样品所含色谱峰较少,有利于溶剂系统筛选,为后续高速逆流色谱分离奠定分离基础。3.2样品f1高速逆流色谱分离对小叶金钱草乙酸乙酯萃取物的分析可知,样品所包含化学成份复杂且极性较大,如想从成分复杂的样品中建立快速有效的高速逆流色谱分离策略难度很大,在粗样中难以确定分离目标。根据常规溶剂体系,如正己烷:乙酸乙酯:甲醇:水、二氯甲烷:甲醇:水以及正己烷:正丁醇:水等溶剂体系来摸索分配系数,或者将溶剂系统中的正己烷替换为石油醚,甲醇替换为乙腈,二氯甲烷替换为氯仿等等。hplc测定化合物在溶剂体系中的分配系数k,k值定义为上相化合物色谱峰面积(s上)除以下相化合物色谱峰面积(s下)(k=s上/s下),k值在0.2~5.0间的溶剂系统用于高速逆流色谱。另一方面,两组份的分离度(α=k1/k2,k1>k2)应该大于1.5。分配系数的测定:将适量的样品粉末溶解在适当量的预平衡的两相溶剂系统中,并置于分液漏斗中。剧烈摇动分液漏斗以彻底平衡两相样品,并取等体积的两相蒸发至干。残余物用1ml乙腈超声溶解,过0.45μm滤膜并按照3.1.2中方法进行hplc分析。参考常见高速逆流色谱溶剂系统,如正己烷:乙酸乙酯:甲醇:水、氯仿:甲醇:水以及正己烷:正丁醇:水等,按照溶剂极性的不同配制体积比为1:1:1:1的正己烷:乙酸乙酯:甲醇:水、2:1:1的二氯甲烷:甲醇:水、1:1:1的乙酸乙酯-正丁醇-水和1:1:1的正己烷-正丁醇-水系统,考察此四个比例系统对样品分配系数k的影响。按照二氯甲烷:甲醇:水的体积比配制溶剂系统,比例依次为:3:1:1、2.5:1:1、2:1:1、1.5:1:1和1:1:1。考察氯仿对k的影响。由实验前期研究可知,色谱峰在二氯甲烷:甲醇:水溶剂系统中上相和下相分布差异较大,而在其余溶剂系统中色谱峰分布差异不显著。综上所述,选用二氯甲烷:甲醇:水溶剂系统分离f1样品,探索合适分配系数,建立快速有效的高速逆流色谱分离方法。当使用hsccc分离样品时,需要考虑一些重要的性质,例如样品的极性是否合适,分配系数是否受温度或浓度的显着影响,等等。k值越小越早流出。随着k值的增加,样品的出峰时间趋于延长,色谱峰比以前更宽,但能与其他样品更好地分离。最重要的是,目标化合物必须是稳定的并且易溶于溶剂体系中,并且溶剂体系能够迅速地分成两相。分配系数见表3.2。部分化合物极性适中,且在乙酸乙酯中溶解性较好。换句话说,大多数化合物总是在含有乙酸乙酯的有机相中富集。基于以上原因,我们选择了不含乙酸乙酯的溶剂系统,选择二氯甲烷/甲醇/水系统。首先,选择二氯甲烷-甲醇-水(2:1:1,v:v)来测试微量化合物的富集。根据溶剂系统给出的k值(2:1:1,v:v),色谱峰1,4能够从复杂的样品系统中分离,而化合物2和3不能实现有效的分离。幸运的是,我们发现通过调节二氯甲烷的比例可以有效地分离化合物2和3,而不影响其他化合物的分离。因此,通过调节溶剂系统中二氯甲烷的比例来调节化合物的分配系数。随着二氯甲烷的增加,化合物2、3k值更接近,即化合物2、3的α值减小,故不能分离。相反,降低二氯甲烷的比例可以提高分离度而不影响其他化合物的分离。此外,二氯甲烷-甲醇-水(1.5:1:1,v:v)系统给出了最高的固定相保留和目标化合物的合适k值,因此选择它用于进一步分离。表3.2f1样品在氯仿-甲醇-水溶剂体系中的分配系数kkkk序号氯仿-甲醇-水(v/v/v)123413:1:15.122.592.983.9422.5:1:14.952.342.763.7232:1:14.731.992.433.4041.5:1:14.671.622.113.1151:1:14.281.481.732.82配制二氯甲烷-甲醇-水(1.5:1:1,v:v)溶剂系统,于分液漏斗中充分振摇后在室温下静置10h,将上下相分离并超声脱气20min,上相为固定相(水、甲醇)下相为流动相(氯仿、甲醇)。以20ml/min流速将上相泵入主机。待主机中充满上相后,启动主机正相旋转按键fwd,调整主机转速为900r/min,仪器转速稳定后再以2ml/min的流速泵入超声脱气后的下相。待流动相流出主机后,平衡数分钟待基线平稳后计算固定相保留率(sf),计算方法为:sf=[(螺旋管体积-流出固定相体积)/螺旋管体积]×100%。开启tbd2000检测系统,设置检测波长为254nm,采集数据。取3.1.3中f1分离样品3g,用20ml上相溶解,由进样阀上样,记录色谱图,收集各分离组分。采用二氯甲烷-甲醇-水(1.5:1:1,v:v)溶剂系统。参照上文固定相保留率计算方法,sf=[(300-95)]/300×100%=68.3%。得到7个分离组分并进行hplc分析,其余高速逆流色谱分离组分的化学成分正在研究中。根据分离经验及hplc初步分析,第一个随流动相流出的色谱峰为杂质峰,一般为极易溶解于流动相的物质,这部分物质的分批系数一般小于0.2,分离价值小、无分离意义,故不做详细研究。最后通过1h、13cnmr从逆流色谱峰中鉴定了其中四个化合物,即化合物1,2,5和7。保留时间及色谱峰面积百分比如下:化合物1,rt=10.752min,92.85%;化合物2,rt=9.627min,97.42%,化合物5,rt=43.625min,94.65%,化合物7,rt=42.710min,95.42%。由紫外光谱分析得知,化合物5和7色谱峰紫外吸收在峰带i:300-400nm及峰带ii:220-280nm有明显紫外吸收,符合黄酮类化合物紫外光谱吸收特征,即化合物5、7为黄酮类化合物。而化合物1和2最大紫外吸收在254nm左右,为简单苯环衍生物天然产物,可能为有机酸类天然产物。3.3样品f2高速逆流色谱分离参考高速逆流色谱分离黄酮类化合物的相关报道选择溶剂系统,配制比例均为2:1:1的乙酸乙酯-正丁醇-水和正己烷-正丁醇-水系统,考察此两个比例系统对分配系数k的影响。按照正己烷-正丁醇-水的体积比配制溶剂系统,比例依次为:1:1:1、1:2:2、1.5:1:1、1.75:1:1、1:2:1、2:2:1和2:3:1。在初步的试验当中,目标化合物极易溶于乙酸乙酯和水。在表3.3中可以看出,1号溶剂乙酸乙酯-正丁醇-水(2:1:1,v:v)分配系数过大,因此不适合作为高速逆流色谱溶剂系统。在2号系统中将乙酸乙酯替换为正己烷后体系的分配系数大为改善,但表3.3中2号系统中1号化合物分配系数较小,会过早的和杂质一同随流动相流出,化合物纯度将无法保证,因此不选用2号溶剂系统。因此下一步通过调整正己烷和正丁醇的比例来探索正己烷-正丁醇-水溶剂系统的最适分配系数。表3.3f2分离样品在不同溶剂体系中的分配系数k溶剂系统比例(v/v/v)分配系数分离度编号乙酸乙酯:正丁醇:水2:1:1k1=72.90;k2=78.671.071正己烷:正丁醇:水2:1:1k1=0.31;k2=0.882.832由表3.3、表3.4可知,2号、3-9号溶剂系统的k值在0.2~5.0之间。在上相作为流动相和下相作为固定相的溶剂体系中,分配系数越小出峰时间越早。通过调整正己烷和正丁醇的比例时可以发现,当增加正己烷或减小正丁醇的比例时,化合物的分配系数减小而增加分离度。由3号及7号、2号及8号和8号及9号可知,增加正丁醇的比例对化合物的分配系数影响较大,即延长逆流色谱出峰时间。更重要的是:由于上样量较大(300mg),为了避免hsccc前后色谱峰发生重叠,综合考虑,通过调整正己烷的比例可以在保证分离度的前提下尽可能的缩短分离时间,节约溶剂。选用分配系数适中的3号溶剂系统正己烷-正丁醇-水(1:1:1,v:v)进行高速逆流色谱分离目标物。表3.4f2分离样品在正己烷-正丁醇-水溶剂体系中的分配系数k比例(v/v/v)分配系数分离度编号1:1:1k1=1.22;k2=3.082.5231:2:2k1=5.12;k2=10.562.0641.5:1:1k1=0.76;k2=2.042.6851.75:1:1k1=0.54;k2=1.542.7561:2:1k1=2.55;k2=4.541.7872:2:1k1=1.09;k2=2.672.4582:3:1k1=2.37;k2=4.701.989配制两相溶剂体系:正己烷-正丁醇-水(1:1:1,v:v)。仪器参数(正相旋转,转速900r/min,流速2ml/min,检测波长为254nm,f2分离样品300mg固定相保留率(sf),sf计算方法同3.2.2。采用正己烷-正丁醇-水(1:1:1,v:v)溶剂系统。参照3.2.2中sf计算方法,sf=[(300-108)]/300×100%=64.0%。按照3.2.2方法步骤进行hsccc分离,得到4个hsccc组分。并将所得各组分进行hplc分析。峰i为混乱杂质峰,没有进一步分离价值。峰ii-iv的hplc分析,经1h和13cnmr鉴定,峰ii(a)、iii(b)和iii(c)分别对应化合物9、10和12,结构鉴定见3.6。保留时间和纯度分别为:化合物9,13.894min,90.12%;化合物10,rt=18.974min,97.85%;化合物12,rt=26.282min,95.42%。由紫外光谱分析得知,色谱峰紫外吸收在峰带i:300-400nm及峰带ii:220-280nm有明显紫外吸收,符合黄酮类化合物紫外光谱吸收特征,故所分离得到的三种化合物为黄酮类化合物。冷冻干燥后分别得8.12mg,118.53mg,127.59mg。3.4样品f3高速逆流色谱分离配制正己烷-正丁醇-水-冰乙酸(2:3:1:0.1,v:v)两相溶剂系统,操作步骤同3.2.2。仪器参数(正相旋转,转速900r/min,流速2ml/min,检测波长为254nm,f3分离样品50mg。采用正己烷-正丁醇-水(2:3:1:0.1,v:v)溶剂系统,sf=[(300-96)]/300×100%=68.0%。进行hsccc分离,得到5个组分进行hplc分析。通过1h、13cnmr鉴定了其中一个化合物,即化合物13,保留时间和纯度分别为:rt=13.874min,87.55%。色谱峰在峰带i:300-400nm及峰带ii:220-280nm有明显紫外吸收,符合黄酮类化合物紫外光谱吸收特征,即黄酮类化合物。冷冻干燥后得4.53mg化合物13。3.5样品f4高速逆流色谱分离根据前期实验总结出的经验,在含有乙酸乙酯的溶剂系统中化合物的分配系数过大,故在f4样品的分离过程中使用不含乙酸乙酯的正己烷-正丁醇-水溶剂系统。按照正己烷-正丁醇-水的体积比配制溶剂系统,比例依次为:2:1:1、1:1:1、1:2:1、1:2:2和1:1:2。按照正己烷-正丁醇-水的体积比配制溶剂系统,比例依次为:2:1:1、1:1:1、1:2:2和1:2:1。由表3.5可知,1、2号溶剂系统的k范围在0.2~5.0之间。而3、4号中化合物的k大于5,故不采纳。通过调整正己烷和正丁醇的比例时可以发现,当增加正己烷或减小正丁醇的比例时,化合物的分配系数减小而增加分离度。且正丁醇的比例对化合物的分配系数影响较大,即延长逆流色谱出峰时间。综合考虑,通过调整正己烷的比例可以在保证分离度的前提下尽可能的缩短分离时间,节约溶剂。选用2号系统进行分离。表3.5样品f4在正己烷-正丁醇-水溶剂体系中的分配系数k比例(v/v/v)分配系数分离度编号2:1:1k1=0.07;k2=0.27;k3=0.343.85;1.2511:1:1k1=0.56;k2=1.55;k3=2.332.98;1.5021:2:2k1=1.60;k2=3.38;k3=6.512.11;1.9231:2:1k1=2.59;k2=5.26;k3=6.522.03;1.234正己烷-正丁醇-水(1:1:1,v:v)两相溶剂系统。仪器参数(正相旋转,转速900r/min,流速2ml/min,检测波长为254nm,f2分离样品40mg)固定相保留率(sf)。正己烷-正丁醇-水(1:1:1,v:v)溶剂系统,sf=[(300-100)]/300×100%=66.1%。得到4个组分并进行hplc分析。峰i为混乱杂质峰,没有进一步分离价值。经1h和13cnmr鉴定为化合物11,保留时间和纯度为:rt=25.733min,90.55%。由紫外光谱分析得知,色谱峰紫外吸收在峰带i:300-400nm及峰带ii:220-280nm有明显紫外吸收,符合黄酮类化合物紫外光谱吸收特征,故所分离得到的化合物为黄酮类化合物。冷冻干燥得15.59mg化合物11。3.6结构解析及鉴定经1hnmr、13cnmr及ms等多种表征手段,鉴定出乙酸乙酯萃取物中14个单体化合物的结构,并对部分化合物列出详细解析过程。从乙酸乙酯萃取物中分离得到的化合物可以分为两类:有机酚酸类和黄酮(苷)。esi-ms/ms分为两种模式:即正离子模式(positive)和负离子模式(negative)。负离子模式化合物分子量变化情况主要有两种情况:[m-h]-和[m+cl]-,即分子量减1或加35.5。正离子模式则较为复杂,出现情况列举如下:[m+h]+、[m+nh4]+、[m+na]+、[2m+h+h]2+和[2m+h+na]2+,化合物分子量相应发生变化。有机酚酸类化合物可以在除苯环之外的部分进行裂解,可以生成三类互补离子,一是取代的苯甲酰基离子,二是通过八元环过渡态转移氢原子,从而形成一对互补离子,三是只能进行官能团的裂解。从小叶金钱草中分离的有机酸类化合物有四种,均为简单苯环取代产物,即香草酸、对羟基苯甲酸、原儿茶酸和没食子酸。简单苯环衍生物属于上述情况的第三类,ms/ms裂解后大多只能发现母体碎片峰而不易发现断裂官能团碎片,对于分离的有机酸类化合物ms/ms裂解在这里不做详细研究。分离得到的简单取代黄酮有四种:槲皮素、山柰酚、木犀草素和芹菜素。简单黄酮为只有羟基或甲氧基取代的黄酮类化合物,ms/ms裂解情况与苷类化合物裂解情况相同,这里不做赘述,与黄酮苷类化合物合并探讨。另外,对于黄酮类化合物的esi-ms中出现[m-1]峰这类基本情况做一探讨,其质谱行为黄酮类化合物当中,[m-1]离子较强,氢离子的来源有可能是b环中的c2’位。化合物1:白色无定形粉末(ch3oh);分子式为c8h8o4;uv(meoh)λmax:260,291nm;esi-ms:m/z169[m+h]+。1hnmr(dmso-d6,500mhz,ppm):δ12.35(1h,s,–cooh),9.93(1h,s,–oh),7.50(1h,dd,j=8.0,2.0hz,6-h),7.49(1h,d,j=2.0hz,2-h),6.89(1h,d,j=8.4hz,5-h),3.81(3h,s,–och3)。13cnmr(dmso-d6,125mhz,ppm):δ167.68(-cooh),151.42(c-3),147.63(c-4),123.87(c-6),122.11(c-1),115.43(c-5),112.19(c-2),55.98(-och3)。结构鉴定过程如下:根据化合物1的1hnmr图谱,首先判断氘代试剂种类:1hnmr在2.55,3.43出现信号,13cnmr在39ppm处出现信号,为dmso-d6的氢谱溶剂峰、水峰以及碳谱溶剂峰。可将其质子信号分为三类。1、芳环质子信号;2、低场区活泼氢信号;3、烷氧基信号。根据图谱可判断化合物1为简单苯环取代产物,根据其积分结果和特征信号可判断包含三种官能团:1、位于低场区的弱宽单峰(brs),根据特征官能团的化学位移可判断其为羧基氢信号,表示为12.35(1h,brs,-cooh);2、同样位于低场区的一组弱宽峰,可判断其为羟基活泼氢质子信号,表示为9.93(1h,s,-oh);3、靠近高场区在3.5~3.9ppm处出现尖锐的单峰,表示为3.81(3h,s,-och3)。而芳环质子信号在7.50,7.49,6.89ppm出现,分别表示为:7.50(1h,d,j=2.0hz,6-h),7.49(1h,s,2-h),6.84(1h,d,j=8.4hz,5-h),其中7.50ppm处质子信号与6.84处质子信号符合向心现象,判定为5,6-h。由于2,6-h与羧基(拉电子基团)临近向低场区位移,故在7.50ppm处信号为6-h,7.49ppm处信号则为2-h。由于5-h与羟基(富电子基团)临近向高场区位移,故在6.84ppm处信号为5-h。根据nmr数据分析,并与已报道文献[43]比对,化合物1鉴定为香草酸(vanillicacid)。化合物2:白色无定形粉末;分子式为c7h6o3;uv(meoh)λmax:254nm;esi-ms:m/z138[m+h]+。1hnmr(dmso-d6,500mhz,ppm):δ12.07(1h,s,-cooh),10.25(1h,s,–oh),7.73(2h,d,j=8.4hz,2,6-h),6.76(2h,d,j=8.4hz,3,5-h)。13cnmr(dmso-d6,125mhz,ppm):δ167.49(-cooh),161.88(c-4),131.92(c-2,6),121.71(c-3,5),115.49(c-1)。根据已报道文献比对,化合物2鉴定为4-羟基苯甲酸(p-hydroxybenzoicacid)。化合物3:白色无定形粉末;分子式为c7h6o4;uv(meoh)λmax:259,293nm;esi-ms:m/z154[m+h]+。1hnmr(dmso-d6,500mhz,ppm):δ10.12(2h,s,-oh),7.34(1h,s,2-h),7.30(1h,d,j=7.55hz,6-h),6.79(1h,d,j=7.50hz,5-h)。13cnmr(dmso-d6,125mhz,ppm):δ167.98(–cooh),150.42(c-4),145.35(c-3),122.42(c-6),121.31(c-1),117.07(c-2),115.62(c-5)。根据已报道文献比对,化合物3鉴定为原儿茶酸(protocatechuicacid)。化合物4:白色无定形粉末;分子式为c7h6o5;uv(meoh)λmax:270nm;esi-ms:m/z170[m+h]+。1hnmr(dmso-d6,500mhz,ppm):δ12.00(1h,s,-cooh),9.15(3h,s,-oh),6.90(1h,s,2,6-h)。13cnmr(dmso-d6,125mhz,ppm):δ167.53(-cooh),145.42(c-3,5),137.99(c-4),120.51(c-1),108.73(c-2,6)。根据已报道文献比对,化合物4鉴定为对没食子酸(gallicacid)。化合物1-4为简单有机酚酸类化合物,其1hnmr较为简单,通过对其氢谱进行研究便可确定结构,首先是信号的耦合裂分情况,通过向心现象确定相关氢的耦合情况,根据耦合常数的大小判断氢组信号为邻位耦合或是间位耦合。其次,最简单的,根据氢信号裂分为d或dd峰判断官能团的取代位次。最后是活泼氢,因为活泼氢较多,因此在进行nmr测试时首选dmso-d6作为测试溶剂,以便研究对其活泼氢进行研究,最常见的就是酚羟基和羧基氢。但nmr中活泼氢信号有以下特点。峰尖锐或较宽,积分经常较小,与其它质子信号重叠,几乎与谱图基线一致。这可能与活泼氢的交换速度有关,交换速度慢的一般表现为宽单峰(brs),速度快的则为尖锐的单峰。化合物5:黄色无定形粉末;分子式为c15h10o7;uv(meoh)λmax:254,364nm;esi-ms:m/z302[m+h]+。1hnmr(dmso-d6,500mhz,ppm):δ12.69(1h,s,5-oh),10.75(1h,brs,7-oh),10.45(1h,brs,4'-oh),7.93(2h,d,j=8.7hz,h-2',6'),6.94(2h,d,j=8.7hz,h-3',5'),6.78(1h,s,h-3),6.49(1h,d,j=2.0hz,h-8),6.21(1h,d,j=2.0hz,h-6)。13cnmr(dmso-d6,125mhz,ppm)δ147.26(c-2),136.20(c-3),176.30(c-4),156.60(c-5),98.65(c-6),164.36(c-7),93.81(c-8),161.19(c-9),103.47(c-10),122.42(c-1'),116.07(c-2'),145.52(c-3'),148.16(c-4'),115.53(c-5'),120.43(c-6')。结构鉴定过程如下:对于图谱中核磁兆数的判断,有种较为简便且实际的方法。偶和常数分为同碳质子耦合(2j)、邻碳质子耦合(3j)和间位耦合/远程耦合(4j),其中3j,其j的范围一般在6~9之间,而间位耦合j值较小,一般在1~3之间。选取两组为二重峰耦合裂分的氢来判断,以槲皮素中两组耦合常数不同的氢为例。根据图谱判断6.90处为邻位耦合,(6.9097-6.8927)*核磁兆数结果应落在6~9之间,显然核磁兆数应为500mhz。同样的根据第二组信号也可判断核磁兆数为500mhz。在黄酮类化合物当中,对于结构的判断很大程度依赖于偶和常数以及通过向心现象判断氢的耦合。在槲皮素(化合物5)的1hnmr图谱中,δh6.20(d,1h,j=2.1)和6.42(d,1h,j=2.1)为黄酮h-6和h-8的特征质子信号(即a环5,7二羟基取代时),从j=2.1判定氢信号为间位耦合。h-6和h-8位的两组信号峰型对称,信号外侧较低而内侧较高,符合向心现象的规律。6.78ppm左右没有出现黄酮3-h信号,判定3号位被羟基取代。通过对信号进行积分可知为三组不同氢信号。对信号分别进行积分和耦合常熟的计算,结果如下:7.70(1h,d,j=2.2),7.56(1h,dd,j=2.2,8.5)和6.90(d,1h,j=8.5)。7.56ppm处出现dd峰,有两组j值:2.2和8.5,表明该氢既与邻位氢耦合又s与间位氢耦合,判定为h-6’。6.90ppm处j=8.5,表明该氢只发生邻位耦合,判定为h-5’。7.70ppm处j=2.2,表明该氢只发生间位耦合,故判定为h-2’。结合低场区活泼氢信号积分之和为5,所以该化合物为3,5,7,3’,4’-五羟基黄酮,即槲皮素,分析结果与文献一致。同样的,根据信号耦合关系也可推断出化合物6、7和8的结构。化合物6:黄色无定形粉末;分子式为c15h10o6;uv(meoh)λmax:264,363nm;esi-ms:m/z286[m+h]+。1hnmr(dmso-d6,500mhz,ppm):δ12.49(1h,s,5-oh),10.77(1h,s,4′-oh),10.13(1h,s,7-oh),9.40(1h,s,3-oh),8.06(2h,d,j=8.9hz,h-2′,h-6′),6.94(2h,d,j=9.0hz,h-3′,h-5′),6.45(1h,d,j=2.1hz,h-8),6.21(1h,d,j=2.1hz,h-6)。13cnmr(dmso-d6,125mhz,ppm)δ:147.28(c-2),136.13(c-3),176.38(c-4),160.19(c-5),98.68(c-6),164.37(c-7),93.95(c-8),156.65(c-9),103.52(c-10),122.15(c-1′),129.98(c-2′,c-6′),115.91(c-3′,c-5′),159.66(c-4′)。根据已报道文献比对,化合物6鉴定为山柰酚(kaempferol)。化合物7:黄色无定形粉末;分子式为c15h10o6;uv(meoh)λmax:252,347nm;esi-ms:m/z286[m+h]+。1hnmr(dmso-d6,500mhz,ppm):δ12.97(1h,s,5-oh),10.81(1h,s,7-oh),9.91(1h,s,3’-oh),9.45(1h,s,4’-oh),6.72(1h,s,h-3),6.24(1h,d,j=2.0hz,h-6),6.49(1h,d,j=2.0hz,h-8),7.45(1h,d,j=2.0hz,h-2'),6.94(1h,d,j=8.2hz,h-5'),7.45(1h,dd,j=8.2,2.0hz,h-6')。根据已报道文献比对,化合物7鉴定为木犀草素(luteolin)。化合物8:黄色无定形粉末;分子式为c15h10o5;uv(meoh)λmax:266,337nm;esi-ms:m/z270[m+h]+。1hnmr(dmso-d6,500mhz,ppm):δ12.49(1h,s,5-oh),10.77(1h,s,4′-oh),10.13(1h,s,7-oh),9.40(1h,s,3-oh),8.06(2h,d,j=8.9hz,h-2′,h-6′),6.94(2h,d,j=9.0hz,h-3′,h-5′),6.45(1h,d,j=2.1hz,h-8),6.21(1h,d,j=2.1hz,h-6)。13c-nmr(dmso-d6,125mhz,ppm):δ164.20(c-2),103.30(c-3),182.21(c-4),161.92(c-5),99.30(c-6),164.60(c-7),94.43(c-8),157.77(c-9,c-4'),104.17(c-10),121.64(c-1'),128.93(c-2',c-6'),116.42(c-3',c-5')。根据已报道文献比对,化合物8鉴定为芹菜素(apigenin)。化合物5-8为简单黄酮类化合物,其1hnmr并不复杂,通过对其氢谱进行研究便可确定结构,重点在于母核结构和官能团的判断。而为了不漏掉活泼氢,nmr测试溶剂同样选择dmso-d6作为测试溶剂。在之前讨论中以详细介绍过nmr测试的溶剂选择及判断方法,这里不再赘述。化合物9-13,16为黄酮苷类化合物,由于化合物16从正丁醇萃取物中也分离得到,故在此不做讨论,归并到正丁醇分离化合物结构鉴定中讨论。与简单黄酮相比,其1hnmr会在5~6ppm处出现一组甚至多组d峰,这类信号为糖苷端基氢的特征信号,对黄酮糖苷的判断提供重要依据。化合物9:黄色无定形粉末;分子式为c21h20o12;uv(meoh)λmax:259,357nm;esi-ms:m/z464[m+h]+。1hnmr(dmso-d6,500mhz,ppm):δ12.69(1h,s,5-oh),10.90(1h,s,7-oh),9.30(2h,s,3′,5′-oh),8.89(1h,s,4′-oh),6.90(2h,s,2′,6′-h),6.37(1h,d,j=1.9hz,8-h),6.20(1h,d,j=1.8hz,6-h),5.12(1h,s,1″-h),3.15-3.99(4h,m,rha-2,3,4,5),0.85(3h,d,j=6.2hz,ch3)。13c-nmr(dmso-d6,125mhz,ppm):δ177.22(c-4),164.90(c-7),161.77(c-5),157.92(c-8a),156.90(c-2),146.25(c-3′,5′),136.96(c-4′),134.72(c-3),120.06(c-1′),108.35(c-2′,6′),104.42(c-4a),102.40(c-1″),99.20(c-6),94.02(c-8),71.74(c-4″),71.02(c-3″),70.85(c-2″),70.48(c-5″),18.00(c-6″)。结构鉴定过程如下:化合物9,根据1hnmr可将其质子信号分为三类:1、低场区活泼氢信号;2、芳环质子信号;3、糖基信号(包括端基氢信号和其他特征信号)。确定为黄酮苷类化合物。化合物主要结构片段:端基信号δh5.12(1h,s)(由于裂分情况不理想,因此端基氢并未裂分成二重峰)和双峰甲基信号δh0.85(3h,d,j=6.2hz)及几个δh3.0~4.0ppm之间的质子信号,证实化合物结构中存在鼠李糖基。化合物10:黄色无定形粉末;分子式为c21h20o11;uv(meoh)λmax:260,349nm;esi-ms:m/z448[m+h]+。1hnmr(dmso-d6,500mhz,ppm):δ12.66(1h,s,5-oh),9.74(3h,brs,7,3′,4′-oh),7.31(1h,d,j=2.0hz,2′-h),7.26(1h,dd,j=2.0hz,j=8.3hz,6′-h),6.87(1h,d,j=8.3hz,5′-h),6.39(1h,d,j=1.7hz,8-h),6.20(1h,d,j=1.8hz,6-h),5.27(1h,s,1″-h),3.14-3.99(4h,m,rha-2,3,4,5),0.83(3h,d,j=6.1hz,ch3)。13c-nmr(dmso-d6,125mhz,ppm):δ178.15(c-4),165.08(c-7),161.75(c-5),157.69(c-2),156.94(c-8a),148.95(c-4′),145.70(c-3′),134.64(c-3),121.57(c-6′),121.17(c-1′),116.08(c-5′),115.92(c-2′),104.40(c-4a),102.28(c-1″),99.26(c-6),94.15(c-8),71.64(c-4″),71.05(c-2″),70.81(c-3″),70.52(c-5″),17.97(c-6″)。根据已报道文献比对,化合物10鉴定为槲皮苷(quercitrin)。化合物11:黄色无定形粉末;分子式为c21h20o11;uv(meoh)λmax:264,347nm;esi-ms:m/z448[m+h]+。1hnmr(dmso-d6,500mhz,ppm):δ12.63(1h,s,5-oh),10.84(1h,s,7-oh),10.20(1h,s,4′-oh),8.05(2h,d,j=8.9hz,2′,6′-h),6.89(2h,d,j=8.9hz,h-3′,5′),6.44(1h,d,j=2.1hz,h-8),6.21(1h,d,j=2.1hz,h-6),5.47(1h,d,j=7.45hz,glc-1),3.08~3.58(6h,m,glc-h)。13c-nmr(dmso-d6,125mhz,ppm):δ177.94(c-4),164.61(c-7),161.70(c-5),160.42(c-4′),156.85(c-8a),156.72(c-2),133.65(c-3),131.37(c-2′,6′),121.37(c-1′),115.58(c-3′,5′),104.48(c-4a),101.31(c-1″),99.16(c-6),94.12(c-8),77.98(c-3″),76.89(c-5″),74.69(c-2″),70.36(c-4″),61.30(c-6″)。根据已报道文献比对,化合物11鉴定为紫云英苷(astragalin)。化合物12:黄色无定形粉末;分子式为c21h20o13;uv(meoh)λmax:254,347nm;esi-ms:m/z480[m+h]+。1hnmr(dmso-d6,500mhz,ppm):δ12.57(1h,s,5-oh),9.00(4h,brs,7,3′,4′,5′-oh),7.14(2h,s,h-2′,6′),6.30(1h,d,j=1.6hz,h-8),6.12(1h,d,j=1.6hz,h-6),5.27(1h,d,j=7.7hz,gal-1),3.20-3.57(6h,m,gal-h)。根据已报道文献比对,化合物12鉴定为杨梅素-3-o-半乳糖苷(myricetin3-o-galactoside)。化合物13:黄色无定形粉末;分子式为c21h20o13;uv(meoh)λmax:258,355nm;esi-ms:m/z480[m+h]+。1hnmr(dmso-d6,500mhz,ppm):δ12.59(1h,s,5-oh),9.16(4h,brs,7,3′,4′,5′-oh),7.13(2h,s,h-2′,6′),6.29(1h,d,j=1.6hz,h-8),6.11(1h,d,j=1.6hz,h-6),5.39(1h,d,j=7.7hz,glc-1),3.02-5.27(6h,m,glc-h)。根据已报道文献比对,化合物13鉴定为异杨梅树皮苷(杨梅素3-o-葡萄糖苷)isomyricitrin(myricetin3-o-glucoside)。乙酸乙酯萃取物分离得到的黄酮苷有6种,分别为:杨梅苷(9)、槲皮苷(10)、紫云英苷(11)、杨梅素3-o-半乳糖苷(12)、异杨梅树皮苷(杨梅素3-o-葡萄糖苷)(13)和槲皮素3,3'-二-α-l-鼠李糖苷(16)化合物16结构鉴定合并至正丁醇分离化合物结构鉴定中。上述黄酮所连接糖苷均为氧苷,而不是碳苷,且连接糖苷均为单糖苷,同一连接位置无二糖苷等多糖苷出现。黄酮氧苷类化合物的主要裂解是由弱的分子离子(单糖苷)失去糖苷恢复原来的黄酮母核结构,恢复母核结构以后在c环发生裂解,之后继续发生裂解失去h2o、co和o。所连接单糖苷有四种:鼠李糖、葡萄糖、半乳糖以及葡萄糖醛酸(单糖名称、分子式、分子量及缩写见表3.6),氧苷键断裂后失去相应的糖残基(失去分子量为单糖分子量-18)。采用sephadexlh-20纯化与高速逆流色谱相结合的方法从乙酸乙酯萃取物中分离纯化出,成功从小叶金钱草中分离出包含同分异构体在内的黄酮苷、酚类等14种单体化合物(香草酸(1)、4-羟基苯甲酸(2)、原儿茶酸(3)、没食子酸(4)、槲皮素(5)、山柰酚(6)、木犀草素(7)、芹菜素(8)、杨梅苷(9)、槲皮苷(10)、紫云英苷(11)、杨梅素3-o-半乳糖苷(12)、异杨梅树皮苷(杨梅素3-o-葡萄糖苷)(13)和槲皮素3,3'-二-α-l-鼠李糖苷(16)),通过核磁共振波谱对有机化合物特征信号进行总结研究及结构鉴定。另一方面,通过二级质谱手段对化合物裂解模式进行研究,为化合物结构解析提供准确的方法。实施例4该实施例提供小叶金钱草化学成分生物活性鉴定方法,包括如下步骤:4.1仪器与试剂raw264.7小鼠单核巨噬细胞、dmem培养基、biotoppeddmso、胰蛋白酶-edta消化液、pbs缓冲液、1%p/s青链霉素混合液、无菌塑料细胞培养瓶、无菌96孔板、一次性无菌过滤器0.22μm,武汉普诺赛生命科技有限公司;新生牛血清,美国gibco公司;地塞米松,美国sigma公司;elisa试剂盒(il-6和tnf-α),南京建成生物工程研究所。伯乐xmark酶标仪上海道尚生物科技有限公司;hf90二氧化碳培养箱力康生物医疗科技控股有限公司;td3低速离心机湘仪离心机;kj-201c微量振荡器江苏康健医疗用品有限公司;mb100-4p微孔板孵育器金坛市盛蓝仪器制造有限公司;fa2004b电子天平上海越平科学仪器有限公司;alrstreami级a2型生物安全柜新加坡esco公司:其他器材试剂:细胞计数板、培养皿、去离子水等4.2主要试剂的配制方法raw264.7完全培养基的配置:dmem高糖基础培养基、10%胎牛血清(fbs)和1%p/s青链霉素混合液按体积比89:10:1混合,过0.22μm一次性无菌过滤器,4c冷藏,备用。elisa试剂盒标准品的配制:(1)tnf-α:按照试剂盒说明书配制1280ng/l,640ng/l,320ng/l,160ng/l,80ng/l和40ng/l的标准品溶液。(2)il-6:按照试剂盒说明书配制320ng/l,160ng/l,80ng/l,40ng/l,20ng/l和10ng/l的标准品溶液。4.3抗炎活性试验使用dmem完全培养基中培育raw264.7,培养瓶置于37℃、5%co2培养箱中培养。对于测试化合物,浓度设定为高剂量组、中剂量组和低剂量组。使用文献报道的方法并做适当修改,用于评估抗炎活性。简言之,将raw264.7细胞接种于96孔板,用不同浓度的化合物(低剂量:5μg/ml,中剂量:50μg/ml,高剂量:500μg/ml)在37℃培养箱中培养1小时,再加入lps(0.1μg/ml)培养24小时。使用商业酶联免疫吸附测定(elisa)试剂盒评估细胞上清液的il-6和tnf-α的产生水平。地塞米松用作阳性对照。所有测试值均以给出。阳性药物组:包含浓度为50μg/ml地塞米松的完全培养基。当p<0.05时,实验结果具有统计学意义。4.4elisa酶联免疫试剂盒数据处理根据试剂盒中标准品,按浓度和od值绘制标准曲线。所得数据通过专用计算软件elisacalc进行计算,拟合模型选用logistic曲线(四参数)。计算方程公式为方程:y=(a-d)/[1+(x/c)^b]+d。4.5elisa酶联免疫试剂盒标准曲线根据配制好的tnf-α和il-6标准溶液测试od值,绘制标准曲线如图5。拟合方程曲线为:tnf-α:y=2.88211/[1+(x/300.86954)^1.54252]-0.10253,r2=0.9987il-6:y=13.9431/[1+(x/0.12946)^0.42268]-0.32348,r2=0.99994.6单体化合物活性测试结果表4.1和4.2是部分单体化合物的对lps诱导下raw264.7巨噬细胞tnf-α和il-6的产生水平的评价。由表4.1可知,与空白对照组相比,lps处理可诱导raw264.7释放大量的炎症因子tnf-α(p<0.05),浓度由98.67±9.34μg/ml上升至1102.64±54.23μg/ml。用不同浓度的单体化合物(低剂量:5μg/ml,中剂量:50μg/ml,高剂量:500μg/ml)预处理raw264.7细胞,则会显著抑制tnf-α的产生。与lps处理组相比,各浓度试验组tnf-α含量均有统计学差异(p<0.05),且具有良好的剂量浓度依赖性。由表4.2可知,与空白对照组相比,lps处理可诱导raw264.7细胞释放大量的炎症因子il-6(p<0.05),浓度由20.53±2.46μg/ml上升至231.52±17.69μg/ml。用不同浓度的单体化合物(低剂量:5μg/ml,中剂量:50μg/ml,高剂量:500μg/ml)预处理raw264.7细胞,则会显著抑制il-6的产生。单体化合物2、3、9、16均具有抑制活性,与lps处理组相比,各浓度试验组il-6含量均有统计学差异(p<0.05),且具有良好的剂量浓度依赖性。经lps诱导培养及化合物处理,细胞上清液中tnf-α和il-6含量均有所降低,与lps诱导组相比差异显著(p<0.05)。单体化合物1、2、7均具有抑制活性,其中,单体化合物2对tnf-α和il-6的产生有较好的抑制作用,在浓度5μg/ml时tnf-α含量为127.57±15.36ng/l,显著低于其它化合物;而测试化合物对il-6的抑制效果整体高于tnf-α,il-6的含量均低于100ng/l。表4.1化合物1,2,和7对lps诱导下raw264.7中tnf-α的产生水平(ng/l)组别127化合物低剂量170.3±20.55*127.57±15.36*231.27±25.18化合物中剂量76.13±6.99*39.47±5.25*164.12±19.63*化合物高剂量24.43±1.29*28.13±1.57*44.49±2.75*空白98.67±9.34lps诱导组1102.64±54.23#地塞米松组127.21±7.19*note:#p<0.05vsblankgroup,*p<0.05vslpsgroup表4.2化合物2,3,9和16对lps诱导下raw264.7中il-6的产生水平(ng/l)note:#p<0.05vsblankgroup,*p<0.05vslpsgroup有益效果:实验验证了lps可以诱导raw264.7细胞释放大量的il-6和tnf-α,而当使用化合物对raw264.7细胞培养后,lps诱导的il-6和tnf-α的产生被明显抑制,说明化合物的抗炎作用是下调lps诱导的细胞因子il-6和tnf-α作用的结果。化合物1、2、3是有机酚酸类化合物,化合物7是简单黄酮类化合物,9、16是黄酮苷类化合物。在高浓度时对tnf-α和il-6的产生均有较好的抑制作用,而化合物2与其它化合物相比,无论在高浓度和低浓度均有比较好的抑制作用。从表中还可以看出,化合物对il-6的抑制效果要高于tnf-α,就活性而言,有机酚酚酸类的活性要高于黄酮类化合物。以上所述仅是本申请的具体实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本申请原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本申请的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1