一种枸橼酸坦度螺酮稳定晶型及其制备方法与流程
1.本发明涉及枸橼酸坦度螺酮的晶型技术领域,具体涉及枸橼酸坦度螺酮的一种稳定晶型及其制备方法。
背景技术:
2.坦度螺酮最早由日本住友制药株式会社研制,于1996年在日本批准上市。坦度螺酮属于氮杂螺酮(azapirone)类药物,为第3代抗焦虑药,作用于5-ht受体,在脑内与5-ht1a受体选择性结合,主要作用部位集中在情感中枢的海马、杏仁核等大脑边缘系统以及投射5-ht能神经的中缝核。药物通过激动5-ht1a自身受体,调节从中缝核投射至海马的5-ht,抑制行动抑制系统的5-ht效应,发挥抗焦虑作用,其结构式如下:
[0003][0004]
苯二氮卓类(bdz)药物主要作用于bdz受体,具有肌肉松弛、镇静、催眠及影响精神运动功能的作用。坦度螺酮与bdz类药物相比,坦度螺酮的结合部位分布相对集中,可发挥选择性更高的抗焦虑作用,基本无镇静、不诱导睡眠、无抗抽搐效应,无肌肉松弛效应,无依赖效应,从而减少了bdz类药物常见的不良反应。枸橼酸坦度螺酮临床上主要用于广泛性焦虑症,如伴发焦虑的高血压患者和伴发焦虑的肠易激综合征患者以及慢性阻塞性肺病(copd)患者焦虑症状的治疗,市场前景非常看好。
[0005]
目前,国外对枸橼酸坦度螺酮晶型未见报道。国内专利如:cn 102344442 a、cn 103641817 a、cn 103641818 a和cn105985327 a等对晶型进行了相关研究。虽然上述专利公开了枸橼酸坦度螺酮的多种晶型,但均为混合晶型且制备方法存在操作繁琐,成本高,环境不友好等缺点。
技术实现要素:
[0006]
鉴于此,本发明提供了一种枸橼酸坦度螺酮的新晶型,以及该晶型的制备方法。
[0007]
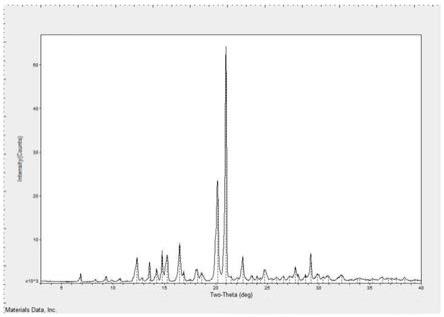
为了实现上述目的本发明采用的技术方案是:一种枸橼酸坦度螺酮稳定晶型,所述晶型的x-射线粉末衍射图谱中,2θ衍射角在:12.3
±
0.2
°
,16.5
±
0.2
°
,20.2
±
0.2
°
,21.0
±
0.2
°
处有特征衍射峰。
[0008]
进一步,晶型的x-射线粉末衍射图谱中,2θ衍射角还在:13.6
±
0.2
°
,14.8
±
0.2
°
,15.3
±
0.2
°
,16.9
±
0.2
°
,18.2
±
0.2
°
,18.6
±
0.2
°
,22.6
±
0.2
°
,24.7
±
0.2
°
,27.7
±
0.2
°
,29.2
±
0.2
°
,32.2
±
0.2
°
处有特征衍射峰。
[0009]
进一步本发明优选方案中,枸橼酸坦度螺酮的晶型,使用cu-kα辐射,具有基本如图1所示的x-射线粉末衍射图。
[0010]
进一步本发明优选方案中,所述晶型具有基本如图2所示的dsc图谱。
[0011]
本发明还提供了一种枸橼酸坦度螺酮稳定晶型的制备方法,包括以下步骤:坦度螺酮游离碱与枸橼酸加入到溶剂中,坦度螺酮游离碱与枸橼酸摩尔比为1:1.0~1:3.0;0~80℃搅拌至析出固体。可以是直接析出固体或降温后析出固体。
[0012]
在上述制备方案中,优选摩尔比为坦度螺酮游离碱:枸橼酸=1:1.0~1:1.5。
[0013]
在上述制备方案中,所述溶剂选自质子性溶剂或非质子极性溶剂中的一种或几种的组合。
[0014]
进一步,所述质子溶剂选自水、甲醇、乙醇和异丙醇中的一种或几种的组合;所述非质子极性溶剂选自丙酮、乙腈、乙酸乙酯中的一种或几种的组合。
[0015]
在上述制备方案中,优选溶剂为:水、甲醇、乙醇、异丙醇和丙酮。
[0016]
在上述制备方案中,还包括如下步骤:将固体从混悬液中分离;将分离的固体干燥得到枸橼酸坦度螺酮,干燥温度为60~150℃;干燥时间为3~24小时。
[0017]
特别地,优选摩尔比为坦度螺酮游离碱:枸橼酸=1:1.2;溶剂为水、甲醇或乙醇,其体积是坦度螺酮游离碱的重量的5~20倍,单位是ml/g。
[0018]
在上述制备方案中,枸橼酸为枸橼酸无水合物或枸橼酸一水合物。
[0019]
本发明公开的枸橼酸坦度螺酮,具有晶型单一、晶型稳定性好、质量稳定的特点。本发明制备方法采用一步成盐法,避免了现有制备方法中的重结晶操作,制备工艺简化,成本降低。本方法使用的有机溶剂单一,溶剂可以回收套用,避免了现有方法中使用混合溶剂不易回收的缺点。另外本发明还开发出使用水做溶剂,做到了既经济又环保。本方法制备的枸橼酸坦度螺酮的纯度可达到99.8%以上。
[0020]
本发明制备的枸橼酸坦度螺酮由于其晶型单一,晶型稳定性好,这些特点,使得其在制备相应药物中具有便于长久储存,药用安全性好的优点。
附图说明
[0021]
图1为实施例1制备的枸橼酸坦度螺酮的xrpd图;
[0022]
图2为实施例1制备的枸橼酸坦度螺酮dsc图;
[0023]
图3为实施例3制备的枸橼酸坦度螺酮的xrpd图;
[0024]
图4为实施例5制备的枸橼酸坦度螺酮的xrpd图;
[0025]
图5为实施例10高湿试验的枸橼酸坦度螺酮xrpd图;
[0026]
图6为实施例10中加速试验6个月的枸橼酸坦度螺酮xrpd图;
具体实施方式
[0027]
本发明的枸橼酸坦度螺酮稳定晶型的结构表征使用了x射线粉末衍射,差热扫描量热法。化合物纯度使用waters高效液相色谱检测。
[0028]
x射线粉末衍射使用高分辨透射模式xrpd图在panalytical x’pert3 x射线粉末衍射分析仪上采集,xrpd测试参数如下:x射线kα11.540598;扫描范围(
°
2th)3
°-
40
°
;扫描步长(
°
2th)0.0131;每步扫描时间(s)97.767。
[0029]
差热扫描量热使用t-a差热热量及dsc2000,操作方法:使用t-zero瓶,加热范围为30~350℃,加热速度为每分钟5℃,氮气流速为80ml/min。
[0030]
下面结合具体的实例对本发明的技术方案作进一步的说明:
[0031]
实施例1枸橼酸坦度螺酮稳定晶型的制备
[0032]
称取坦度螺酮游离碱100.0g,枸橼酸50.1g,加入甲醇1500ml,搅拌升温至回流,缓慢降温至0~20℃,保温搅拌2小时,过滤,滤饼使用甲醇洗涤,得枸橼酸坦度螺酮湿品。将湿品于65℃干燥15小时,得枸橼酸坦度螺酮,收率为88.1%,纯度99.89%,其xrpd如图1,dsc如图2。
[0033]
实施例2枸橼酸坦度螺酮稳定晶型的制备
[0034]
称取坦度螺酮游离碱100.0g,枸橼酸60.1g,加入甲醇1500ml,室温搅拌至固体析出,再搅拌2小时,过滤,滤饼使用甲醇洗涤,得枸橼酸坦度螺酮湿品。将湿品于65℃干燥15小时,得枸橼酸坦度螺酮,收率为91.4%,纯度99.82%。
[0035]
实施例3枸橼酸坦度螺酮稳定晶型的制备
[0036]
称取坦度螺酮游离碱100.0g,枸橼酸60.1g,加入乙醇1500ml,搅拌升温至回流,缓慢降温至0~20℃,保温搅拌2小时,过滤,滤饼使用乙醇洗涤,得枸橼酸坦度螺酮湿品。将湿品于80℃干燥10小时,得枸橼酸坦度螺酮,收率为95.8%,纯度99.87%,其xrpd如图3。
[0037]
实施例4枸橼酸坦度螺酮稳定晶型的制备
[0038]
称取坦度螺酮游离碱100.0g,枸橼酸75.1g,加入纯化水500ml,搅拌升温至60~80℃,降温至0~20℃,保温搅拌2小时,过滤,滤饼使用水洗涤,得枸橼酸坦度螺酮湿品。将湿品于90℃,干燥12小时,得枸橼酸坦度螺酮,收率为92.3%,纯度99.82%。
[0039]
实施例5枸橼酸坦度螺酮稳定晶型的制备
[0040]
称取坦度螺酮游离碱100.0g,枸橼酸75.1g,加入纯化水500ml,室温搅拌至固体析出,再搅拌2小时,过滤,滤饼使用水洗涤,得枸橼酸坦度螺酮湿品。将湿品于90℃干燥12小时,得枸橼酸坦度螺酮,收率为90.6%,纯度99.86%,其xrpd如图4。
[0041]
实施例6枸橼酸坦度螺酮稳定晶型的制备
[0042]
称取坦度螺酮游离碱100.0g,枸橼酸100.2g,加入丙酮2000ml,搅拌升温至回流,降温至0~20℃,保温搅拌2小时,过滤,滤饼使用丙酮洗涤,得枸橼酸坦度螺酮湿品。将湿品于65℃,干燥15小时,得枸橼酸坦度螺酮,收率为96.2%,纯度99.84%。
[0043]
实施例7枸橼酸坦度螺酮稳定晶型的制备
[0044]
称取坦度螺酮游离碱100.0g,枸橼酸60.1g,加入乙腈2000ml,搅拌升温至回流,降温至0~20℃,保温搅拌2小时,过滤,滤饼使用乙腈洗涤,得枸橼酸坦度螺酮湿品。将湿品于75℃,干燥12小时,得枸橼酸坦度螺酮,收率为93.0%,纯度99.81%。
[0045]
实施例8枸橼酸坦度螺酮稳定晶型的制备
[0046]
称取坦度螺酮游离碱100.0g,枸橼酸65.1g,加入异丙醇1500ml,室温搅拌至固体析出,再搅拌2小时,过滤,滤饼使用异丙醇洗涤,得枸橼酸坦度螺酮湿品。将湿品于85℃,干燥8小时,得枸橼酸坦度螺酮,收率为94.8%,纯度99.83%。
[0047]
实施例9枸橼酸坦度螺酮稳定晶型的制备
[0048]
称取坦度螺酮游离碱100.0g,枸橼酸75.1g,加入乙酸乙酯1500ml,室温搅拌至固体析出,再搅拌2小时,过滤,滤饼使用乙酸乙酯洗涤,得枸橼酸坦度螺酮湿品。将湿品于80℃,干燥10小时,得枸橼酸坦度螺酮,收率为95.5%,纯度99.84%。
[0049]
实施例10枸橼酸坦度螺酮的晶型稳定性评价
[0050]
将制得的枸橼酸坦度螺酮晶体开展影响因素试验、加速稳定性试验,试验方法参
见《中国药典(2015)》第二部附录xixc《原料药与药物制剂稳定性试验指导原则》。
[0051]
(一)影响因素试验:
[0052]
1.高温试验:取上述实施例中制备的枸橼酸坦度螺酮,于60℃放置10天,于第5天和第10天取样,测定各指标与0天进行对照,试验结果见表1。
[0053]
2.高湿试验:取上述实施例中制备的枸橼酸坦度螺酮,于湿度在75%下放置10天,于第5天和第10天取样,测定各指标与0天进行对照,试验结果见表1。
[0054]
3.强光照射试验:取上述实施例中制备的枸橼酸坦度螺酮,于照度为(4500
±
500)lux的条件下放置10天,于第5天和第10天取样,测定各指标与0天进行对照,试验结果见表1。选取部分xrpd图谱如图5所示。
[0055]
表1枸橼酸坦度螺酮影响因素试验
[0056][0057]
(二)加速稳定性试验:
[0058]
上述实施例中制备的枸橼酸坦度螺酮在恒温恒湿箱中进行6个月的加速稳定性试验。试验条件湿:40℃/75%相对湿度(rh),分别于0、1、2、3、6个月取样,进行纯度,有关物质和xrpd检测,结果见表2。选取部分xrpd图谱如图6所示。
[0059]
表2枸橼酸坦度螺酮的加速稳定性试验
[0060][0061]
由上述影响因素试验和加速试验可以看出,本发明制备的枸橼酸坦度螺酮在高温、高湿和光照条件下杂质未见明显增加,纯度无明显变化,晶型未发生转变;加速稳定性试验证明产品质量稳定性良好。
[0062]
综上所述,本发明公开的枸橼酸坦度螺酮晶型纯度高且稳定,质量稳定性好,有利于运输,储存以及药物制剂的开发、使用,提高了产品的药用安全性。本发明公开的枸橼酸坦度螺酮晶型的制备方法,具有操作简单、经济环保、成本低廉、适合工业化生产等优点。
[0063]
本发明制备的枸橼酸坦度螺酮晶型可用于枸橼酸坦度螺酮固体制剂的开发,包括与枸橼酸坦度螺酮相关的药品。
[0064]
应当说明的是,以上所述仅为本发明的较佳实施例而已,并不用于限制本发明的范围,凡在本发明的精神和原则之内所作出的任何修改,等同的替换代替和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1