一种合成利托那韦的新方法与流程
本发明涉及药物化学领域,具体涉及一种合成利托那韦的新方法。
背景技术:
利托那韦至今为止报道的合成方法不多,主要的合成方法是通过两个关键中间体结构,n-((n-甲基-n-((2-异丙基-4-噻唑基)甲基)氨基)甲酰)-l-缬氨酸和(2s,3s,5s)-5-(叔丁氧基甲酰胺基)-2-(n-5-噻唑基-甲氧羰基)氨基-1,6-二苯基-3-羟基己烷进行缩合制备。两个片段结构如下:
1.1n-((n-甲基-n-((2-异丙基-4-噻唑基)甲基)氨基)甲酰)-l-缬氨酸甲酯(羧酸中间体)和1.2(2s,3s,5s)-5-氨基-2-(n-5-噻唑基-甲氧羰基)氨基-1,6-二苯基-3-羟基己烷(氨基中间体),两个片段的缩合方式现有的工艺报道如下:
1)原研公司雅培wo9414436专利,该专利使用1-乙基-(3-二甲基氨基丙基)碳酰二亚胺(edc)为缩合剂,1-羟基苯并三唑(hobt)作为弱碱条件进行缩合;该方法使用试剂edc和hobt价格昂贵,生产成本高。反应试剂引入副产物较多,后处理纯化比较繁杂;不利于保证产品质量。
原研公司雅培另一专利us5567823,报道中使用氯甲酸异丁酯和n-羟基丁二酰亚胺在n-甲基吗啉条件下进行缩合。使用的氯甲酸异丁酯和n-羟基丁二酰亚胺价格相对比较高,并且该方法产物收率只有70%,工业生产成本较高,反应过程对体系水分要求较高,工业生产条件要求比较高。
2)东北制药专利cn106749084和cn106749085,分别报道用2-(7-氮杂苯并三氮唑)-n,n,n’,n’-四甲基脲六氟磷酸盐(hatu)和二异丙基碳二亚胺(dic)作为缩合剂进行缩合反应。反应溶剂氯仿毒性大,不利于生产的安全性;试剂丁酮和乙酸丁酯不是常用溶剂,增加工业化大生产的困难。反应副产物四甲基脲和含磷盐类等,后处理纯化比较繁琐,不利于保证产品质量。
总之,合成利托那韦的工艺存在反应试剂价格昂贵,反应收率不高,不利于工艺大生产成本控制;反应条件比较苛刻,操作繁琐,不利于产品质量的控制等缺点。
技术实现要素:
在化学原料药生产领域,原本在实验室研究阶段,小试工艺阶段或中式工艺阶段可行的合成方法,在工业化放大生产时经常出现问题,如杂质含量超标,纯度降低,产率降低等;另外,在实验室研究阶段,小试工艺阶段或中式工艺阶段可用的试剂,在工业化生产中不一定可行,这种例子在药物原料药制备领域比比皆是。在工业化制备合成利托那韦的过程中,也存在同样的问题,现有技术报道的制备方法并不适合工业化生产。
因此,本发明所要解决的技术问题是提供一种适合于工业化生产的合成利托那韦的新方法。
为了解决现有技术存在的问题,本发明采用如下技术方案:
一种合成利托那韦的新方法,
其包括:-5至10℃下,将特戊酰氯加入中间体v中,再滴入有机碱,滴毕,反应釜内温度保持-5至10℃,搅拌反应t1时间后加入4-二甲氨基吡啶,再搅拌t2时间,最后加入中间体vi的二氯甲烷溶液,加完,将反应温度升至25至35℃,搅拌至反应完毕。
在一些实施例中,所述有机碱选自三乙胺,n,n-二异丙基乙胺(diepa),n-甲基吗啉,吡啶和1,8-二氮杂二环十一碳-7-烯(dbu)等有机碱中的一种或者多种混合;在一些实施方案中,所述有机碱为三乙胺。
在一些实施例中,所述t1时间为0.5小时以上,优选1小时以上,在某些实施例中为1-2小时。在一些实施例中,所述t2时间0.5小时。
本发明的优点:
反应中采用特戊酰氯、4-二甲氨基吡啶、二氯甲烷和有机碱组合,可以确保中间体v和特戊酰氯反应活化阶段,羧酸活化完全;将特戊酰氯加入中间体v中,再滴入有机碱、4-二甲氨基吡啶最后加入中间体vi的二氯甲烷溶液,本发明发现加料顺序也严重影响反应的产率和纯度。另外,物料加入过程中选择适宜的间隔时间t1和t2时间再加入其他物料也有助于提高反应的产率和纯度。由于采用以上技术方案,本发明取得如下有益效果:
1)改用常用工业原料,试剂,可适用于工业化大生产;
2)反应试剂副产物常温下为液态,后处理及产品纯化容易,产品质量能很好的得到控制;
3)反应原料试剂价格合适,有利于生产成本的控制;
4)反应条件简单,操作方便,有利于大生产产能的提高;
5)经过简单纯化获得产率85%以上,纯度99.7%以上。
附图说明
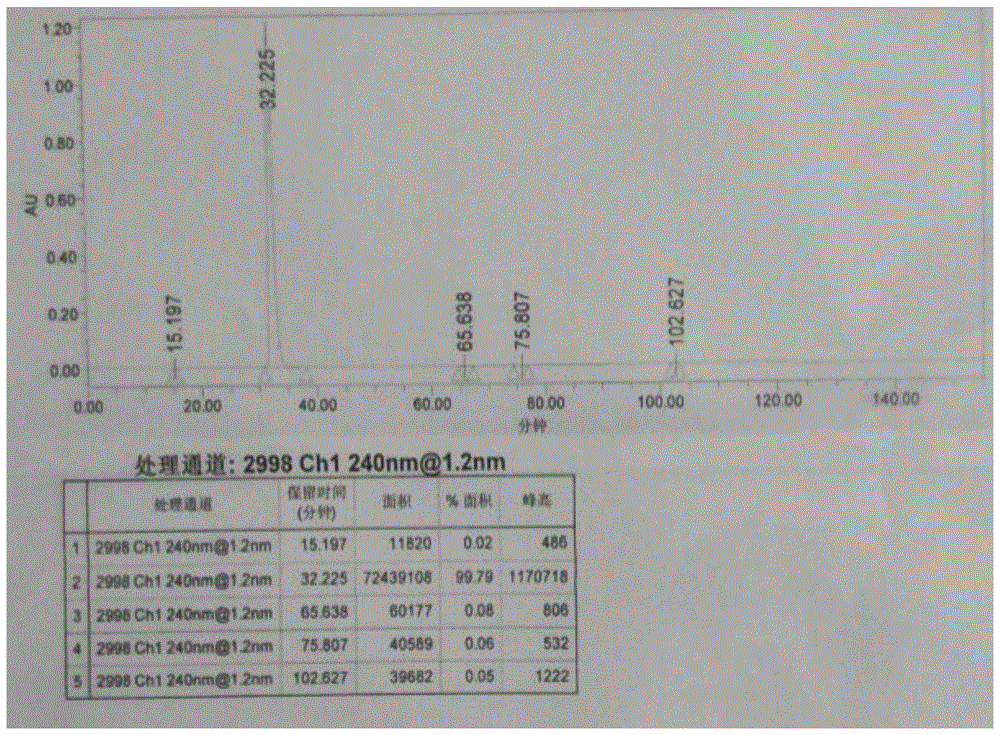
图1示本发明实施例1制备的利托那韦成品的高效液相检测图谱。
具体实施方式
为了使本领域的技术人员更好地理解本发明的技术方案,下面进一步披露一些非限制实施例对本发明作进一步的详细说明。
实施例1一种合成利托那韦的新方法
反应釜r101抽真空,氮气破空,氮气保护下将二氯甲烷1500kg,中间体v共165kg加入釜内,开启搅拌降温至5±5℃;将特戊酰氯58.0kg加入釜内;加完,保持反应釜r101温度5±5℃,通过高位罐v101-i将三乙胺50.0g滴加入釜内;滴毕,反应釜内温度保持5±5℃,搅拌反应1~2小时;往釜内加入12.0kg的4-二甲氨基吡啶(dmap),搅拌30分钟,然后加入中间体vi的二氯甲烷溶液(183.0g溶于500kg);加完,将釜内温度升至30±5℃,搅拌反应8~10小时;取样,hplc检测中间体vi反应完全。
利托那韦反应液的后处理方法
保持反应釜内温度20±5℃,缓慢加入5%氢氧化钠水溶液(500kg),搅拌30分钟,静置30分钟;下层有机相转移至反应釜r102,上层水层转移至存储罐;
r102釜内加入5%氢氧化钠水溶液(500kg),搅拌30分钟,静置30分钟,下层有机相转移至反应釜r101,上层水层合并至存储罐;
r101釜内加入1m的稀盐酸溶液(60kg工业盐酸溶于500kg水),搅拌30分钟,静置30分钟,分出水层;下层有机相转移至反应釜r103;
保持反应釜r103内温<50℃,开启减压蒸馏回收二氯甲烷至基本无馏分;
反应釜r103加入乙酸乙酯180kg,继续保持反应釜r102内温<50℃,减压蒸馏乙酸乙酯至基本无馏分;
反应釜r103加入乙酸乙酯1000.0kg,将釜内温度升至45±5℃搅拌至完全溶解,然后加入无水硫酸钠100kg和活性炭20kg,釜内温度升至65±5℃,搅拌脱色30分钟;
反应釜r103内物料保温65±5℃通过压滤器过滤,乙酸乙酯80.0kg洗涤r103釜壁和滤饼,滤液一并转移至洁净区内反应釜r201中。
保持反应釜r201内温65±5℃,加入正庚烷800kg,保温搅拌30分钟;
将反应釜r201内温在4~5小时内降至35±5℃,析出固体,保温搅拌1小时;
继续将反应釜r201内温降至15±5℃,保温搅拌1小时;
将反应釜r201内物料放入离心机甩滤,23kg乙酸乙酯和17kg正庚烷混合液共40kg淋洗;
固体湿品放入双锥内,控制温度50±5℃减压烘干,降温至25±5℃出料包装得到成品利托那韦260kg~280kg,产率85%以上,纯度99.795%,详见附图1。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!