慢病毒感染人表皮角质细胞株及其构建方法和应用与流程
本发明属于分子生物学、基因工程以及皮肤基础科学领域研究,具体涉及慢病毒感染人表皮角质细胞株及其构建方法和应用。
背景技术:
:角质形成细胞是构成表皮的主要细胞,占到表皮细胞80%以上。研究发现,表皮角质细胞的增殖过度在许多恶性皮肤疾病包括银屑病中均有发生。诸如:砷中毒及其引起的皮肤癌前期出现表皮细胞过度增殖以及皮肤异常角质化。银屑病的临床特征同样表现为表皮角质细胞过度增生,表皮角质形成细胞异常增殖与大量鳞屑的产生相关。目前针对银屑病的药物治疗手段主要有调节角质形成细胞异常增殖和分化、免疫抑制剂、阻断t细胞活化和阻断中性粒细胞趋化药物,然而,以上药物治疗手段疗效均较差,且各有显著的副作用,因此迫切需要进一步探寻参与银屑病发生发展重要的环节、发掘更好的靶分子及其适当的抑制途径。磷酸化修饰是调节蛋白活性、稳定性的重要生物事件,对于维持机体生理活动具有重要意义。机体蛋白的磷酸化稳态调控主要是由各种磷酸酶和激酶共同调节。丝苏氨酸磷酸酶2a(pp2a)是人体细胞中占比最高的磷酸酶,主要是针对特定的ser/thr进行去磷酸化的蛋白酶,在真核细胞中大部分的ser/thr位点的去磷酸化是由pp2a所催化的。目前的研究已经证明pp2a参与多种蛋白的合成、信号通路的传导、细胞代谢、细胞周期以及细胞骨架的形成等生命活动,更多的研究表明pp2a在肿瘤的发生和转移过程中发挥重要的作用,通常被作为一种抑癌基因进行研究。pp2a全酶是由结构亚基a(65kd)、调节亚基b(50-130kd)和催化亚基c(36kd)构成的异三聚体。pp2a催化亚基c又具有2种亚型,分别是pp2acα(由ppp2ca基因编码)和β(由ppp2cb基因编码)。c亚基的α亚型占比远高于β亚型(约9倍以上),ppp2ca基因具有维持pp2a全酶活性的重要作用。在肿瘤以及退行性神经疾病的相关研究中发现:pp2a在细胞的增殖、凋亡以及细胞命运的调控中扮演重要的角色,基于pp2a维持机体磷酸化稳态的重要功能,pp2a在其他疾病中的研究仍需要更加深入。针对在许多严重恶性皮肤疾病比如:皮肤癌、银屑病等,表皮角质细胞细胞的异常增殖、凋亡、细胞周期的紊乱是一个典型的病理特征,未来此领域仍有需要更加深入的研究。基于pp2a在调控细胞命运中所扮演的重要角色,结合目前pp2a在皮肤中的研究仅少量存在于临床报道和动物模型,缺乏离体细胞模型进行深入研究的现状。技术实现要素:发明目的:为了解决
背景技术:
中存在的问题,本发明填补了此在皮肤角质细胞中研究ppp2ca功能的细胞模型缺失的空白,不仅针对银屑病相关研究,与异常增殖相关的其他皮肤疾病探寻其疾病发生发展重要的环节、发掘更好的靶分子及其适当的抑制途径同样具有较好的现实意义,本发明为进一步深入研究pp2a在皮肤疾病和未来药物研发提供有力保障。本发明以慢病毒载体的形式表达短发卡rna,通过rna聚合酶ⅲ启动子转录其反向重复dna序列进而形成小发卡结构,通过dicer切割成小干扰rna,从而对靶基因产生持续稳定的特异性沉默作用。本发明中所使用的慢病毒(lentivirus)相比逆转录病毒或者腺病毒等其他病毒载体,除了可转到分裂细胞的优势以外,同时还可以转到非分裂细胞,进而将外源基因整合到靶细胞基因组中,于rna干扰技术集合从而实现对于靶基因的稳定、持续的沉默。本发明以shrna慢病毒为基础构建ppp2ca敲低的人表皮角质细胞株,通过观察发现感染细胞株细胞增殖水平能力显著增高,为探讨ppp2ca在表皮细胞功能作用和作为表皮细胞异常增殖疾病临床治疗靶点提供前期实验数据和研究材料。本发明所要解决的技术问题是提供一种shrna片段。本发明还要解决的技术问题是提供了一种含有所述的shrna片段的重组质粒。本发明还要解决的技术问题是提供了一种重组菌株。本发明还要解决的问题是提供了重组质粒的构建方法。本发明最后要解决的问题是提供了重组菌株的构建方法。本发明涉及慢病毒感染人表皮角质细胞株(hacat)敲减内源性基因,并成功建立并稳筛细胞株。通过应用rnai技术,利用ppp2ca-shrna感染hacat细胞,建立ppp2ca敲减的hacat细胞株,再利用qrt-pcr、westernblot技术对细胞株进行检测,证明细胞株建立成功。技术方案:为了解决上述技术问题,本发明提供了一种shrna片段,所述shrna片段序列如seqidno:1或seqidno:2所示。本
发明内容还包括一种含有所述的shrna片段的重组质粒。其中,所述重组质粒是将shrna片段与质粒pgmlv-sc5相连得到。本
发明内容还包括所述的shrna片段、所述的重组质粒在基因敲低方面的应用。本
发明内容还包括一种重组菌株,所述重组菌株含有所述的shrna片段或所述的重组质粒。本
发明内容还包括重组质粒的构建方法,包括以下步骤:1)设计并合成如所述的shrna片段;2)shrna的退火得到目标片段;3)shrna载体的酶切及回收;4)将步骤3)的酶切载体与步骤2)的目标片段连接得到重组载体。本
发明内容还包括所述的重组菌株的构建方法,包括以下步骤:将所述的重组质粒导入到宿主细胞中得到。其中,本发明所述宿主细胞包括但不仅限于hacat细胞。其中,所述的重组菌株的构建方法,所述具体步骤为:1)构建含有所述的shrna片段的重组表达载体;2)将步骤1)获得的重组表达载体感染宿主细胞hacat获得感染细胞株;3)将步骤2)获得的感染细胞株应用嘌呤霉素筛选培养基定时更换培养基并长期筛选培养,获得阳性细胞株。其中,所述步骤3)的含有嘌呤霉素的筛选培养基为含有2ug/ml的嘌呤霉素的10%的胎牛血清dmem培养基。本
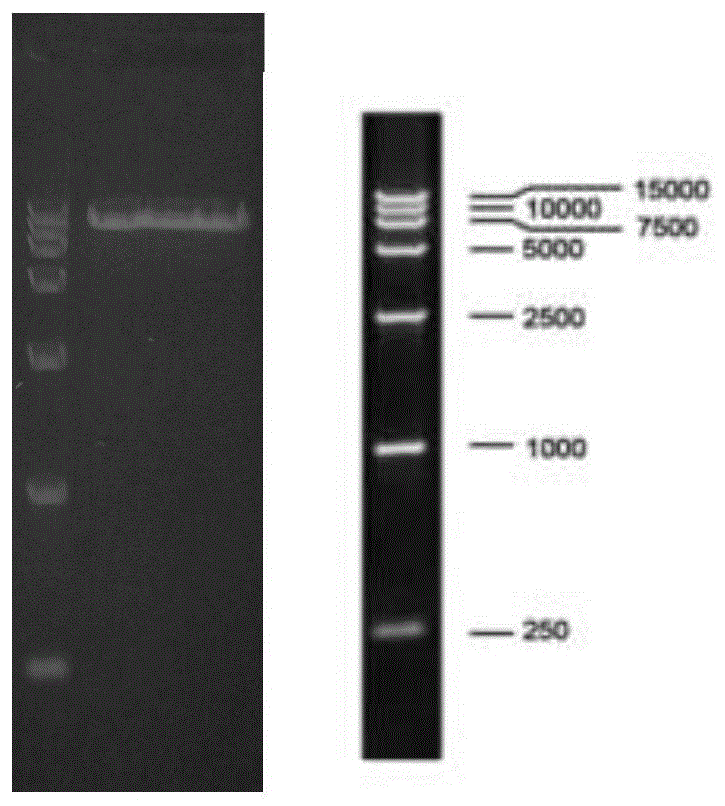
发明内容还包括所述的shrna片段、所述的重组质粒、所述的重组菌株在细胞增殖方面或药物筛选方面的应用。本发明后续将筛选获得的阳性细胞株提取rna和蛋白,通过qrt-pcr和westernblot技、cck-8以及克隆形成实验检测阳性细胞株及其对照组。其结果显示:相比对照组,重组细胞株ppp2ca在mrna和蛋白水平表达显著降低,细胞增殖能力显著增加。有益效果:相对于现有技术,本发明具有以下优点:1、本发明的细胞株遗传稳定性强,经过30次以上传代仍保留转入基因不发生丢失;2、本发明对于ppp2ca的敲低效果显著,可以有效降低其在mrna和蛋白水平的表达;3、本发明细胞株具有良好的实验表型,阳性细胞株的增殖能力显著高于对照组细胞株。附图说明:图1重组载体的酶切电泳图;泳道1:dl15000dnaladder泳道2:digestedwithbamhiandecori;图2、建立ppp2ca敲低的细胞荧光显微图;图中control为对照组慢病毒ppp2ca-nc(sc5),sh1为慢病毒ppp2ca-sh1(sc5)、sh2为慢病毒ppp2ca-sh2(sc5);图3、rt-pcr法对细胞株中的ppp2ca进行检测图;图4、westernblot法对细胞株中的ppp2ca进行检测图;图5、cck8实验检测细胞增殖能力图;图6、克隆形成实验检测ppp2ca敲低细胞增殖能力图;图7、pgmlv-sc5工具载体结构示意图。具体实施方式:下述非限制性实施例可以使本领域的普通技术人员更全面的理解本发明,但不以任何方式限制发明;shrna慢病毒工具载体pgmlv-sc5:购买于上海吉满生物技术有限公司;引物合成:金斯瑞生物科技有限公司dmem培养基:美国hyclone公司;0.05%蛋白酶:美国thermo公司;胎牛血清:美国hyclone公司;嘌呤霉素;中国碧云天公司hgtransgenereagent:上海吉满生物技术有限公司cck8试剂盒:日本同仁pp2acα抗体:美国圣克鲁兹β-actin抗体:美国thermo逆转录试剂盒:日本takara实时定量pcr试剂盒:日本takara实施例1ppp2ca-shrna慢病毒载体的构建1、shrna序列的设计针对ppp2ca目的基因靶基因序列,利用block-ittmrnaidesigner网站进行rna干扰序列设计,设计结果https://rnaidesigner.thermofisher.com/rnaiexpress/design.do我们选择了accessionnumber为cr457417.1,orf区域为1-930,start位点分别为483(shrna1)和217(shrna2)的序列作为shrnaoligos。对照组nc片段为无意义通用序列,且不靶向目标基因。设计的shrna1~2序列和对照nc序列如下:no.targetseqncttctccgaacgtgtcacgtshrna1gcagatcttctgtctacatggshrna2ggcaaatcaccagatacaaat2、shrna引物的设计根据以上基因序列分别设计并合成shrna寡聚单链dna的上下游引物序列,引物序列见下表:3、rnai慢病毒重组质粒的构建将寡聚单链dna退火成双链,将双链的shrnaoligo分别插入到shrna慢病毒工具载体pgmlv-sc5中,构建shrna慢病毒重组质粒,并转化至感受态细胞dh5α。1)shrna的退火:用无菌的tebuffer稀释引物使终浓度为100μm。分别吸取10μl的步骤2中设计并合成的上游和下游引物混合并吹打均匀放入pcr管内进行退火,程序如下:95℃30s72℃2min37℃2min25℃2min结束后置于冰上放置几分钟得到目标片段可直接连接或于-20℃冷冻保存。2)shrna载体的酶切及回收(载体为同一批次提前切好)酶切体系如下:pgmlv-sc55μg10*buffer5μlbamhi2μlecori2μlddh2o补足50μl于37℃酶切约30min。在此期间,可配置0.8%的琼脂糖凝胶,待酶切结束后进行核酸电泳。电泳结束后切下包含目的片段的胶条。用天平称量总重量并减去空管的重量算出凝胶的重量,按100mg约为100μl来计算凝胶的体积,并加入1倍凝胶体积的bingingsolution置于65℃水浴锅内将凝胶彻底融化。期间适当摇晃ep管,加快凝胶的溶解。将上述液体全部转移到滤柱内,13000转离心30s(可重复一次)。然后弃掉管内液体,向柱内加入500μl的wasolution,13000转离心30s。弃掉管内液体,再向柱内加入500μl的washsolution,13000转离心30s(可重复一次)。然后空离3min。将滤柱放置在一个新的1.5mlep管中,室温晾干。最后在柱子内加入35μl的ddh2o,静置5min,13000转离心1.5min。为了提高回收率,可将溶解的dna再次加入柱子内离心一分钟。弃掉柱子,即为回收的载体片段,并测定浓度16.7ng/μl。酶切电泳图参见图1。3)shrna载体与目标片段的连接连接体系如下:分别于25℃水浴中孵育30min得到连接产物。4)转化将感受态细胞置于冰上待其自然解冻后,把连接产物全部加入感受态细胞中,于冰上放置20min,后于42℃水浴中热击90s。然后迅速置于冰上放置2-3min。加入1000μl不含抗生素的lb培养基于37℃,150rpm振荡培养45min。3000rpm离心2min,弃掉约850μl的上清液,将管底的菌液吹打散开,加入到含有氨苄霉素抗性的培养皿中,用灭菌的涂布器涂匀,倒置于37℃恒温培养箱内过夜培养。5)重组质粒的制备挑取若干个单菌落,进行小量摇菌培养,提取质粒获得pgmlv-sc5-ppp2ca-shrna1慢病毒载体、pgmlv-sc5-ppp2ca-shrna2慢病毒载体以及nc对照组载体。6)测序鉴定阳性克隆。pgmlv-sc5-ppp2ca-shrna1干扰载体的测序结果如下:pgmlv-sc5-ppp2ca-shrna2干扰载体的测序结果如下:经过比对,重组克隆中插入片段序列与设计的oligo序列完全一致,因此慢病毒载体构建成功。4、慢病毒包装以及滴度测定1)293t细胞分盘转染前一天,将已经长好的细胞以合适的比例传代到10cm培养皿中,当细胞长到70%~80%时准备转染。2)转染前换液转染前1~2h将需要转染的细胞换新鲜的培养基,12ml/10cm皿,注意:293t细胞贴壁性不是很好,换液时应小心滴加尽量避免冲起细胞。3)转染取无菌的1.5mlep管或15ml离心管,转染体系按下表:混匀后,室温放置15min-20min后,均匀滴加到提前换过液的培养皿中,后置于co2培养箱中培养。4)加enhancingbuffer转染10~12h后,均匀滴加100×enhancingbuffer促进转染,120μl/皿。5)换液转染18~20h后,小心吸掉细胞培养液弃于盛有消毒液的废液杯中(注意:此时培养基中已含有少量的病毒,必须经处理后才能丢弃,所用的移液枪头等必须经消毒液浸泡至少15min后才能丢弃),然后加15ml新鲜的细胞培养基(或是无血清的dmem或是含血清的dmem,具体的按客户要求)继续培养。6)病毒收集换液48h后,吸取细胞上清液于50ml离心管,4℃,4500g离心5min,上清液用0.45μm滤器过滤后转移到新的离心管中,最后将滤液分批转移到浓缩装置中,4℃,4500g,离心10min,弃下层的液体于盛有消毒液的废液杯中,最后一次4℃,4500g,离心20min,此时可见滤器上层中的液体即为病毒浓缩液,命名为慢病毒ppp2ca-sh1(sc5)、ppp2ca-sh2(sc5)以及对照组慢病毒ppp2ca-nc(sc5)。7)病毒分装与保存将病毒分装后保存于-80℃。8)病毒滴度测定将hek293t细胞培养至对数生长期,病毒稀释用培养液为含10%fbs的细胞培养基。第一天,细胞胰酶消化计数后,按照每孔8000细胞接种96孔板,37度培养过夜,感染时细胞长至30~50%的融合密度;第二天,当天转染时,将保存于-80度冰箱中病毒液冰浴融化,用含有10%fbs的细胞培养液进行梯度稀释:1号稀释液:10μl病毒液+90μl病毒稀释用培养基;2号稀释液:10μl1号稀释液+90μl病毒稀释用培养基;3号稀释液:10μl2号稀释液+90μl病毒稀释用培养基;4号稀释液:10μl3号稀释液+90μl病毒稀释用培养基;5号稀释液:10μl4号稀释液+90μl病毒稀释用培养基;按照以上方式依次配制稀释液。选取所需的细胞孔,吸去90μl培养基,丢弃,轻轻混匀各管慢病毒稀释液,取90μl加入每孔细胞中,放入37℃的细胞培养箱中过夜培养;第三天,去除含慢病毒的培养基,加入100μl的完全培养基;第五天,在荧光显微镜下观察各孔中荧光细胞数量,病毒滴度为表达荧光的细胞数乘以相应稀释倍数。综上,滴度如下表慢病毒名称滴度(tu/ml)ppp2ca-sh1(sc5)6×108ppp2ca-sh2(sc5)6×108ppp2ca-nc(sc5)5×108实施例2ppp2ca-shrna慢病毒感染hacat细胞以及筛选1、将处于对数期生长的人表皮角质hacat细胞以1×105个细胞/ml的浓度接种于48孔板中,10%胎牛血清dmem培养基培养,37℃,5%co2培养箱过夜培养24h,感染时细胞融合度40%左右;2、稀释病毒,感染用慢病毒ppp2ca-sh1(sc5)、ppp2ca-sh2(sc5)以及对照组慢病毒ppp2ca-nc(sc5)最终以moi值20稀释后,感染细胞,铺板细胞取出,弃掉旧培养基,加入稀释病毒液200ml/孔,10%胎牛血清dmem培养及培养,37℃,5%co2培养箱培养8h更换为含10%血清和1%双抗(青霉素和链霉素)的dmem培养基;3、感染48h后,观察绿色荧光亮度情况;4、gfp阳性细胞株加入2μg/ml嘌呤霉素,10%胎牛血清dmem筛选培养基,2-3天更换一次,连续培养2个月;5、降低筛选培养基中嘌呤霉素浓度为1μg/ml,继续培养,保持抗性,进行下一步分子生物学鉴定实验。实施例3利用qrt-pcr检测感染细胞mrna水平ppp2ca的表达情况1、rna浓度和纯度测定:提取实施例2中培养的慢病毒ppp2ca-sh1(sc5)、ppp2ca-sh2(sc5)以及对照组慢病毒ppp2ca-nc(sc5)的rna样本用depc水稀释50倍后,检测od值和rna浓度,od值控制在1.8-2.0之间。2、逆转录pcr反应(日本takara逆转录试剂盒):逆转录反应(20μl体系):①取进口无rna酶pcr管,依次加入rna(1μg)、oligodt1μl、无rna水补足至10μl;②70℃置于pcr仪中反应5min,后立即取出至于冰上至少放置1min;③加入:rnase抑制剂1μl、5×rtbuffer5μl、dntps5μl、逆转录酶1μl;④反应pcr管置于pcr仪中,42℃60min,70℃5min,4℃1min;3、qrt-pcr反应:取进口pcr96孔板,置于冰上进行如下操作,根据qrt-pcr说明书加入mix10μl、上下游引物各0.4μl(引物序列:ppp2ca-f:5′-ggtcaagagcctctgcgagaa-3′ppp2ca-r:5′-ccggtcatggcaccagttat-3′;gaphd-f:5′-gcaccgtcaaggctgagaac-3′;gapdh-r:5′-tggtgaagacgccagtgga-3′),步骤2中获得的逆转录模板cdna1μl、无rna酶水补足至20μl配比配置反应液,根据需求加入于96孔板中。pcr反应条件:第一阶段:95℃×5min,1个循环;第二阶段:95℃×10s、55℃×10s,72℃×10s,45个循环;第三阶段:95℃×5s、65℃×1min、95℃持续,1个循环;第四阶段:4℃×30s,1个循环。反应结束后,对结果使用2-δδt法进行分析。图3为qrt-pcr检测慢病毒感染后hacat细胞中ppp2ca的表达,结果显示:ppp2ca-shrna感染组细胞中的ppp2ca表达低于对照组,ppp2ca表达量相比对照组显著性降低。以上结果证明shrna1、shrna2均可以有效降低ppp2ca在表皮细胞中的表达。首次提出ppp2ca的干扰序列shrna1和shrna2为首次提出其敲减作用并可以有效抑制ppp2ca的基因表达。实施例4利用westernblot检测感染细胞蛋白水平ppp2ca的表达水平实施例2中制备的慢病毒ppp2ca-sh1(sc5)、ppp2ca-sh2(sc5)以及对照组慢病毒ppp2ca-nc(sc5)分别用pbs冲洗,每孔加入200μl裂解液(碧云天ripa变性裂解液),刮板收集置离心管种,得到细胞全蛋白,bca法检测蛋白浓度。10%sds-page聚丙烯酰胺凝胶电泳,转膜,5%脱脂奶粉室温封闭1h,一抗孵育过夜,室温tbst洗pvdf膜,含hpr标记的二抗孵育室温1h,tbst洗膜3次,每次10min,ecl显色。凝胶成像系统曝光,记录图片。图4为westernblot检测ppp2ca-shrna1慢病毒载体和ppp2ca-shrna2慢病毒载体感染后hacat细胞中ppp2ca的表达,shrna1和shrna2感染细胞中的ppp2ca表达低于对照组,表明ppp2ca敲减的hacat细胞株建立成功,可用于其他实验研究。实施例5cck8实验检测细胞增殖能力利用cck8(同仁牌细胞增殖-毒性检测试剂盒)检测感染细胞的增殖水平。分组培养实施例2中的慢病毒ppp2ca-sh1(sc5)、ppp2ca-sh2(sc5)以及对照组慢病毒ppp2ca-nc(sc5),当以上细胞生长至80%左右融合时,胰酶消化,收集贴壁细胞,制备细胞悬液,细胞计数,96孔板每孔加入100μl悬液,置于培养箱中培养,分别于1-6天,每天相同时间向每孔加入10μlcck8溶液,置于培养箱继续孵育2h;酶标仪记录450nm处吸光度;每次实验设置6个复孔,重复3次,根据吸光度统计细胞增殖水平,图5中结果显示从第2天开始慢病毒ppp2ca-sh1(sc5)和ppp2ca-sh2(sc5)的ppp2ca敲减hacat细胞增殖水平相比对照组显著提高(p<0.05)。实施例6克隆形成实验检测ppp2ca敲低细胞增殖能力分组培养实施例2中的慢病毒ppp2ca-sh1(sc5)、ppp2ca-sh2(sc5)以及对照组慢病毒ppp2ca-nc(sc5),当细胞生长至80%左右融合时,胰酶消化,以上3组细胞分别接种500个细胞/孔到6孔培养板中,种细胞接种3孔,十字轻轻晃动培养板,使细胞分散均匀,37℃、5%co2培养箱中培养2周。当出现明显肉眼可见的细胞克隆时,终止培养,弃培养基pbs清洗2次,空气干燥;加入甲醛固定15分钟,弃去甲醛,空气干燥;用浓度0.1%结晶紫染色20分钟,流水缓慢清洗掉结晶紫染料;在显微镜下对形成克隆计数(≥50个细胞为一个克隆),平板克隆形成率=(克隆数目/接种细胞数目)×100%,实验重复3次。图6中结果显示:慢病毒ppp2ca-sh1(sc5)和ppp2ca-sh2(sc5)的ppp2ca敲减hacat细胞克隆形成数量显著高于对照nc组,进一步证明ppp2ca在hacat细胞中的敲减可以促进其增殖。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1