一种病毒形式的新型冠状病毒核酸标准物质及其制备方法与流程
本发明涉及一种病毒形式的新型冠状病毒核酸标准物质及其制备方法,属于生物检测技术领域。
背景技术:
核酸(dna或rna)是病毒的遗传物质,任何物种都有独一无二的核酸序列,所以对新型冠状病毒(sars-cov-2)的特征序列进行核酸检测,就可以知道是否感染了新冠病毒。荧光定量pcr技术是目前进行病毒核酸定量检测的主要技术。检测步骤主要包含从病毒中提取基因片段,rna逆转录为dna,dna扩增放大和核酸含量读取。
荧光定量pcr技术的直接测量结果是ct值,即随着检测目标基因的循环复制,每个反应管内的荧光信号累积到达设定的域值时所经历的pcr反应循环数。每个核酸模板的ct值与该模板的起始拷贝数的对数存在线性关系,起始拷贝数越多,ct值越小。想要将ct值转换成被测核酸样品的拷贝数,就需要与被测核酸片段一样的标准物质,标准物质中被测核酸片段的含量是已知的、准确的。利用已知起始拷贝数的标准物质作出标准曲线,其中横坐标代表起始拷贝数的对数,纵坐标代ct值。因此,只要通过pcr检测获得被测核酸样品的ct值,即可从标准曲线上计算出该样品的起始拷贝数。所以,新型冠状病毒核酸标准物质是用pcr技术测量新冠病毒核酸拷贝数的必要工具,也是核酸检测试剂盒研发和生产过程中准确定值所必需的。此外,该标准物质也可以作为阳性参考品与被测样品进行平行操作,以控制病毒核酸检测结果的准确性。
目前,用于新型冠状病毒核酸检测的标准物质仅有中国计量科学研究院发布的高低两种浓度水平的新型冠状病毒核酸标准物质(编号:gbw(e)091089和gbw(e)091090),和上海市计量院发布的2种新型冠状病毒体外转录rna标准物质(编号:gbw(e)091111和gbw(e)091112)。上述标准物质均为rna形式。现有的核酸标准物质均为核酸形式。用核酸形式的标准物质进行rt-pcr检测的步骤是反转录(rt)、核酸扩增(pcr)以及ct结果读取。可以看出,相较于病毒的核酸pcr检测来说,由于裸露的核酸没有病毒结构,其检测过程缺少了从病毒种提取核酸这一步骤,导致核酸形式的标准物质无法参与病毒检测的每一步骤,从而无法进行病毒核酸检测全过程的标准化和质量控制。
技术实现要素:
本发明所要解决的技术问题是克服现有技术的缺陷,提供一种病毒形式的新型冠状病毒核酸标准物质及其制备方法,该标准物质是以病毒为载体,包裹新型冠状病毒核酸检测的靶标基因,具有病毒拟似结构和新型冠状病毒的特异性核酸序列,可以作为新型冠状病毒的标准品实现rna的绝对定量,即将荧光定量pcr测量结果ct值转换为拷贝数,并且可以参与从病毒核酸提取到pcr结果读取的全部检测步骤,控制核酸检测结果的准确性。
为解决上述技术问题,本发明提供一种病毒形式的新型冠状病毒核酸标准物质,所述标准物质以病毒为载体,包裹新型冠状病毒核酸检测的靶标基因。
优选地,所述靶标基因为orf1ab、n基因和e基因。
优选地,所述病毒载体还包裹内参基因hrnasep基因。
本发明还提供一种病毒形式的新型冠状病毒核酸标准物质的制备方法,包括:
合成靶标基因;
将靶标基因分别载入病毒质粒载体;
将病毒质粒转化入大肠杆菌进行培养;
分离、回收病毒质粒;
将病毒质粒转染hek293t细胞并进行病毒包装;
将包装好的病毒颗粒提纯、重悬后,加冻干保护剂分装,经真空冷冻干燥后封装。
优选地,所述靶标基因的合成方法为:
对新型冠状病毒的开放阅读框orf1ab基因、核衣壳n基因、包膜e基因和内参基因hrnasep基因序列进行修改,使其在5'端和3'端包含随机的终止密码子;
合成上述4种基因并做基因融合,合成4种融合基因片段,分别是orf1ab基因和hrnasep基因、e基因和hrnasep基因、n基因和hrnasep基因以及单独的hrnasep基因。
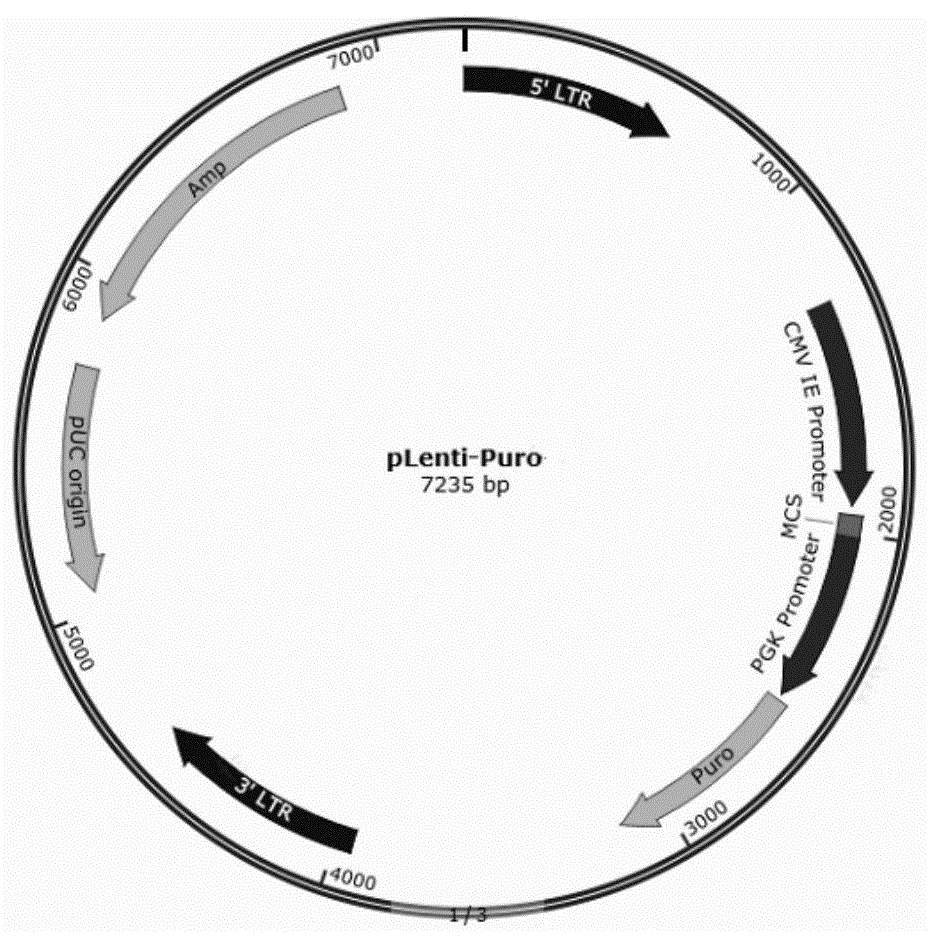
优选地,将靶标基因分别载入病毒质粒载体的方法为:采用genbuildertm克隆试剂盒,将靶标基因分别反向克隆到慢病毒质粒plenti-puro载体中的puromycin抗性基因上游的多克隆位点,同时替换慢病毒质粒载体上的cmv启动子。
优选地,将病毒质粒转化入大肠杆菌的方法为:将病毒质粒分别转化入大肠杆菌stbl3,在lb培养基37℃、14~16小时过夜培养。
优选地,分离、回收病毒质粒的方法为:用碱性裂解提取法分离质粒,经离子交换柱层析、te洗脱、质粒纯化后回收病毒质粒并去除内毒素。
优选地,将病毒质粒转染hek293t细胞及病毒包装的过程为:将病毒颗粒在培养皿中和表达病毒结构蛋白的辅助质粒混合vsvg蛋白、jaj-pol蛋白、rev蛋白,室温孵育20分钟后,转染dmem培养基中的hek293t细胞,37℃、5%co2培养,实现靶标基因片段的转录、病毒外壳蛋白的表达和病毒组装,组装好的病毒分泌胞外。
优选地,所述真空冷冻干燥的方法为:
初步冷冻:将产物在-50℃下保持5小时,然后在-50℃下在0.1mbar下再保持1小时;
初步干燥:在1小时内将温度升至-20℃,并在-20℃下保持30小时;
二次干燥:在10个小时内将温度从-20℃升至25℃,在25℃、0.03mbar下保持15小时。
本发明所达到的有益效果:
(1)本发明的新型冠状病毒核酸标准物质最终以病毒形式呈现,此点是与现有新型冠状病毒核酸标准物质相比的主要区别和优势。现有标物均是rna核酸形式,只能参与目的基因核酸检测的rt-pcr过程。而病毒形式的标准物质与临床样本中的病毒具有相似的生物结构,可以模拟临床病毒样本,进行病毒内核酸提取以及rt-pcr操作,可以控制病毒核酸检测中从核酸提取到核酸定量检测的全过程。
(2)本发明的标准物质中,伴随着新冠病毒特异性基因(orf1ab和n基因、e基因)同时载入了hrnasep基因作为阳性内参对照,可用于监测核酸样品模板的质量、rna提取效率以及pcr的反应效率。hrnasep基因pcr检测失败可能表明从病毒中提取核酸的操作不当,导致rna模板丢失,或反应体系被rt-pcr抑制剂污染。
(3)相较于低温冷冻溶液的保存形式,本发明中运用的真空冷冻干燥技术制备的新型冠状病毒核酸标准物质性能更加稳定,也更适应运输和储存环境。
附图说明
图1是慢病毒质粒载体plenti-puro序列结构图;
图2是分别载入4种靶标基因片段的慢病毒质粒载体结构图;
图3是一种病毒形式的新型冠状病毒核酸标准物质的制备流程图。
具体实施方式
下面对本发明作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。
如图3所示,一种病毒形式的新型冠状病毒核酸标准物质的制备方法,包括以下步骤:
一、构建包含新型冠状毒基因的慢病毒载体
sars-cov-2核苷酸序列来自ncbigenbank登录号nc_045512.2。对sars-cov-2的开放阅读框orf1ab基因、核衣壳n基因、包膜e基因和内标基因人核糖核酸酶p(hrnasep)基因序列进行修改,使其在5'端和3'端包含随机的终止密码子,防止基因表达。基因合成上述4种靶标基因并做基因融合,合成4种融合基因片段,分别是orf1ab基因(部分)和hrnasep基因、e基因和hrnasep基因、n基因和hrnasep基因以及单独的hrnasep基因。采用genbuildertm高效无缝克隆试剂盒,将4种融合基因片段分别反向克隆到慢病毒质粒plenti-puro载体(结构图见图1)中的puromycin抗性基因上游的多克隆位点(mcs),同时替换慢病毒质粒载体上的cmv启动子。每个质粒的最终序列通过sanger测序确认载入基因序列的正确性。将4种慢病毒质粒(结构图见图2)分别转化大肠杆菌stbl3,在lb培养基37℃、14~16小时过夜培养,用碱性裂解提取法分离质粒,经离子交换柱层析、te洗脱、质粒纯化后回收慢病毒质粒并去除内毒素。对回收的质粒进行内毒素检测,小于5eu/mg后即可作为转染级别质粒进入下步使用。
二、慢病毒的包装
上述构建的4种慢病毒颗粒在10cm培养皿中和表达病毒结构蛋白的辅助质粒混合vsvg蛋白、jaj-pol蛋白、rev蛋白,室温孵育20分钟后,转染dmem培养基中的hek293t细胞,37℃、5%co2培养。实现靶标基因片段的转录、病毒外壳蛋白的表达和病毒组装,组装好的病毒分泌胞外。转染后48和72小时收集上清液。上清液经0.45um膜过滤,4℃下转速23000rpm超离心2小时。用冷的dpbs将底部沉淀重悬。采用p24酶联免疫吸附法测定其滴度。使用mycoalerttmplus支原体检测试剂盒检测定支原体,阴性即为合格。
三、标准物质的分装和真空冷冻干燥处理
将上述dpbs缓冲液重悬的4个慢病毒颗粒溶液与适量冻干保护剂充分混匀,冻干保护剂由10mmtris-hcl、ph7.4、0.5%人血清白蛋白,0.1%d-(+)-海藻糖脱水物组成。上述溶液以1ml等分至无菌的5ml安瓿瓶中,虚盖上胶塞,然后将安瓿瓶放入预冻的托盘,装载到真空冷冻干燥器中。初步冷冻,将产物在-50℃下保持5小时,然后在-50℃下在0.1mbar下再保持1小时;初步干燥,在1小时内将温度升至-20℃,并在-20℃下保持30小时。二次干燥,10个小时内将温度从-20℃升至25℃,在25℃、0.03mbar下保持15小时。在冷冻干燥结束时,将容器用干燥氮气回填,并将胶塞完全塞入安瓿瓶中。后续用铝盖封口。
四、标准物质的定值及性能评价:
1.用ddrt-pcr(微滴式数字逆转录-聚合酶链反应)方法对4种标准物质的靶标基因(orf1ab基因、n基因、e基因和hrnasep基因)进行绝对定量(拷贝数/ml),并评定均匀性、稳定性、不确定度。采用的引物和探针序列见表1。
表1ddpr-pcr反应中各靶标基因的引物和探针序列
2.病毒形式的新型冠状病毒核酸标准物质的性能评估
(1)均匀性评估
抽取30个病毒形式的新型冠状病毒核酸标准物质,采用核酸提取试剂盒提取病毒样品的核酸。使用数字pcr方法进行检测,每个样品检测3次,计算平均值。根据抽样方法和检测次数,使用单因素方差分析方法对新型冠状病毒核酸标准物质进行均匀性评估。
(2)长期稳定性评估(即在规定贮存条件下的稳定性)
抽取不少于2瓶的新型冠状病毒核酸标准物质,在-70℃保存0-6个月。采用核酸提取试剂盒提取病毒样品的核酸。使用数字pcr方法进行检测,每个样品检测3次,计算平均值。根据抽样方法和检测次数,使用回归对新型冠状病毒核酸标准物质进行长期稳定性评估。
(3)短期稳定性评估(即在运输过程中的稳定性)
抽取不少于2瓶的新型冠状病毒核酸标准物质,分别在-20℃和4℃1个月、7天或1天。采用核酸提取试剂盒提取病毒样品的核酸。使用数字pcr方法进行检测,每个样品检测3次,计算平均值。根据抽样方法和检测次数,使用回归对新型冠状病毒核酸标准物质进行短期稳定性评估。
3.病毒形式的新型冠状病毒核酸标准物质的定值
将上述方法制备的均匀性高且稳定的新型冠状病毒核酸标准物质进行定值;定值表示为标准值±扩展不确定度。
(1)新型冠状病毒核酸标准物质的标准值
取上述均匀性高且稳定的病毒形式的新型冠状病毒核酸标准物质,由多个独立实验室使用数字pcr方法测定一系列核酸标准物质的特征值,对数据进行统计处理,合作确定标准值。
多个实验室合作定值,当采用同一种方法时,独立定值实验室数不少于8家。
所述核酸标准物质的特征值,是指对于4个核酸标准物质,每个物质重复3次,提供12个特征值。
统计学方法:采用t检验法进行组内可疑值检验,采用f检验法对各组数据的标准偏差进行组间等精度检验,对有显著性差异的数据组在进行技术审查后再决定是否予以剔除。
当各组数据等精度时,检验各组数据平均值是否有显著性差异。如平均值无显著性差异,先合并数据再用适当的方法检验数据分布的正态性,在符合正态分布的情况下,可将两个或多个平均值再次计算平均值,求出总平均值,即为标准值。
4.新型冠状病毒核酸标准物质的扩展不确定度
使用统计学方法计算新型冠状病毒核酸标准物质的扩展不确定度,包括均匀性评估中引入的不确定度、短期和长期稳定性评估中引入的不确定度和定值过程中引入的不确定度。
(1)均匀性评估引入的不确定度的评定
均匀性评估引入的不确定度包括试剂盒的精密度、仪器的精密度和人员操作所引入的不确定度,为a类不确定度。其中由核酸标准物质均匀性产生的标准偏差sbb为:
所以均匀性评估引入的不确定度为:ubb=sbb
(2)稳定性评估引入的不确定度的评定:
稳定性评估引入的不确定度也涵盖了储存温度的变动性对核酸标准物质的影响、试剂盒稳定性引入的不确定度和人员操作引入的不确定度,为a类不确定度。
在产品进行稳定性评估时,对保存在(-20±5)℃环境中的样本,每个月检测一次。每次检测结束后,对数据进行处理,计算其斜率、截距和不确定度。暂定产品有效期为6个月,则有效期t=6个月的稳定性不确定度贡献为:
us=s(β1)·x
(3)定值过程引入的不确定度的评定
不确定度a类评定通过测量数据的标准偏差、测量次数及所要求的置信水平按统计学计算方法进行。由定值测量重复性引入的不确定度u(a)由测量的相对标准偏差计算:
(4)标准值的不确定度的评定
合成上述核酸标准物质的不确定度分量,取扩展因子k=2,计算扩展不确定度。核酸标准物质合成标准不确定度计算公式如下:
标准值的扩展不确定度:ucrm=k×ucrm。
本发明旨在制备一种病毒形式的新型冠状病毒核酸标准物质,本发明中运用的是慢病毒载体包装新型冠状病毒核酸检测的三个靶标基因(n基因、e基因和orf1ab基因),也可以用其他假病毒载体替代本发明的慢病毒载体;也可以用新型冠状病毒核酸检测的其他靶标基因替代本发明中的n基因、e基因和orf1ab基因。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变形,这些改进和变形也应视为本发明的保护范围。
序列表
<110>南京市计量监督检测院
<120>一种病毒形式的新型冠状病毒核酸标准物质及其制备方法
<160>12
<170>siposequencelisting1.0
<210>1
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>1
ccctgtgggttttacacttaa21
<210>2
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>2
acgattgtgcatcagctga19
<210>3
<211>28
<212>dna
<213>人工序列(artificialsequence)
<400>3
ccgtctgcggtatgtggaaaggttatgg28
<210>4
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>4
ggggaacttctcctgctagaat22
<210>5
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>5
cagacattttgctctcaagctg22
<210>6
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>6
ttgctgctgcttgacagatt20
<210>7
<211>26
<212>dna
<213>人工序列(artificialsequence)
<400>7
acaggtacgttaatagttaatagcgt26
<210>8
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>8
atattgcagcagtacgcacaca22
<210>9
<211>26
<212>dna
<213>人工序列(artificialsequence)
<400>9
acactagccatccttactgcgcttcg26
<210>10
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>10
agatttggacctgcgagcg19
<210>11
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>11
gagcggctgtctccacaagt20
<210>12
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>12
ttctgacctgaaggctctgcgcg23
- 还没有人留言评论。精彩留言会获得点赞!