一种快速鉴定二倍体基因编辑作物T0代基因型的方法与流程
本发明属于基因型鉴定技术领域,具体涉及一种快速鉴定二倍体基因编辑作物t0代基因型的方法。
背景技术:
基因组编辑技术在作物育种中得到迅猛发展,这些基因编辑序列特异性核酸酶为基础,包括锌指核酸酶(zincfingernuclease,zfn)、转录激活样效应物核酸酶(transcriptionactivator-likeeffectornucleases,talens)、rna引导的规律成簇间隔短回文重复(clusteredregularlyinterspacedshortpalindromicrepeats,crispr)/cas9及通过crispr/cas9改进的crispr/cpf系统,在基因组特定位点切割双链dna,然后通过非同源末端连接或同源重组方式修复双链缺口,从而在编辑位点附近发生碱基的缺失、插入或替换,达到编辑基因的目的。
植物基因编辑技术往往借助常用的植物遗传转化方法,例如电穿孔、农杆菌介导的遗传转化等方法,对植物基因组特定位点进行插入、删除、替换等编辑,从而快速得到特定目标性状的品种,加快育种速度。但由于基因编辑技术和遗传转化方法的限制,在遗传转化过程中往往产生很少双等位基因纯合突变植物,大部分为杂合体、嵌合体、双等位基因突变、阴性等基因型植株,这对快速鉴定和检测纯合编辑植株带来了挑战。目前鉴定基因编辑植株的方法主要为pcr/re(restrictionendonuclease)方法、错配切割法、taqman方法、高分辨率溶解曲线分析方法,临界退火温度act-pcr(atcriticaltemperaturepcr)、sanger测序等,但是这些方法并不能很好鉴定出t0代的基因型。
技术实现要素:
有鉴于此,本发明的目的在于一种快速鉴定二倍体基因编辑作物t0代基因型的方法,简便快捷,可简单获知基因编辑作物t0代植株的基因型。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种快速鉴定二倍体基因编辑作物t0代基因型的方法,包括以下步骤:(1)以二倍体基因编辑作物t0代的基因组dna为模板dna,在基因编辑靶标位点的上下游设计特异性引物,进行pcr扩增,将得到的pcr产物回收后纯化和第一次sanger测序;当测序测序结果为单一峰图,则所述基因编辑作物t0代的植株为纯合植株(双等位基因纯合突变或纯合的转基因阴性植株);当测序结果单一峰后面出现3个信号峰,则为嵌合体;单一峰后面出现2个信号峰,则为杂合体或双等位基因突变基因型;
(2)对于测序结果有杂峰或套峰的所述pcr产物进行t/a克隆,得连接产物;
(3)将所述连接产物转化感受态细胞,在lb培养基中震荡培养60min后,将菌液涂抹在含氨苄青霉素的lb平板上,培养8~12h,挑取单菌落于含氨苄青霉素的lb培养基中震荡培养8h后,进行第二次sanger测序;将测序结果用ncbiblast工具,与野生型序列进行比对;基因编辑序列为三个不同的序列,则为嵌合体;基因编辑序列为两个不同的序列,则为双等位基因突变基因型;对应的两个序列,一个为野生型,一个为突变序列,则为杂合体基因型。
优选的,步骤(1)中在基因编辑靶标位点的下游350~500bp处设计特异性引物。
优选的,在得到所述特异性引物后,还包括利用所述特异性引物和阴性植株dna进行pcr扩增,扩增条带单一;回收和纯化pcr产物后,进行第一次sanger测序,测序结果显示无杂峰。
优选的,步骤(1)所述pcr扩增的体系以20μl计,包括:模板dna1μl,10mmdntps0.5μl,所述特异性引物的上下游引物各0.5μl,10×extaqbuffer2μl,extaq0.1μl,加水补足至20μl。
优选的,步骤(1)所述pcr扩增的反应程序包括:94℃预变性2min;94℃变性30s,56℃退火50s,72℃延伸40s,35个循环;72℃延伸5min。
优选的,测序结果仅为单一峰图,判定所述基因编辑作物t0代的植株为纯合植株,包括双等位基因纯合突变或阴性植株后,还包括用ncbiblast与野生型靶位点序列进行比对,来判断纯合基因型,如果和野生型序列一致,则为纯合阴性植株;如果不一致,则为双等位基因纯合突变。
优选的,步骤(2)对杂峰pcr产物进行t/a克隆,进行第二次sanger测序;将测序结果用ncbiblast工具与野生型序列进行比对,确定基因编辑的具体序列,验证首次sanger测序峰图对嵌合体基因型的判定,确定杂合体和双等位基因突变基因型:首次sanger测序峰图单一峰后面出现3个信号峰,则所述基因编辑作物t0代的植株为嵌合体,对应的基因编辑序列为三个不同的序列;首次sanger测序峰图,单一峰后面出现2个信号峰,所述基因编辑作物t0代的植株为杂合体或双等位基因突变基因型,和野生型序列相比,如果对应两个序列为不同的序列,则为双等位基因突变基因型,如果对应的两个序列,一个为野生型,一个为突变序列,则为杂合体基因型。
优选的,所述t/a克隆的体系以10μl计,包括:ptopo-bluntsimplevector1μl,pcr产物1μl,10×enhancer1μl和灭菌水7μl。
优选的,步骤(3)所述含氨苄青霉素的lb平板和含氨苄青霉素的lb培养基中氨苄青霉素的浓度均为100μg/ml。
优选的,步骤(3)所述震荡培养和培养的温度均为37℃。
本发明提供了一种快速鉴定二倍体基因编辑作物t0代基因型的方法,首先根据t0代初次sanger测序峰图性质,针对测序结果仅为单一峰图的t0代植株,结合序列比对,确定基因编辑作物的基因型是双等位基因纯合突变或转基因阴性植株(野生型植株);当上述pcr产物的测序结果为杂峰或套峰,根据单一峰后面出现的信号峰的个数,判定具体杂合基因型:单一峰后面出现3个信号峰,则所述基因编辑作物t0代的植株为嵌合体;单一峰后面出现2个信号峰,则所述基因编辑作物t0代的植株为杂合体或双等位基因突变基因型。为了验证对嵌合体基因型的判断及区分杂合体或双等位基因突变基因型,对杂峰pcr产物进行t/a克隆,进行第二次sanger测序;将测序结果用ncbiblast工具与野生型序列进行比对,确定基因编辑的具体序列,验证首次sanger测序峰图对嵌合体基因型的判定,确定杂合体和双等位基因突变基因型,从而实现了快速鉴定基因编辑作物t0代所有基因型的目的。在本发明实施例中,选择二倍体水稻植株通过crispr/cas9技术,得到的loc_os11g12740(ossp1)与loc_os10g41780(oscao1)基因碱基插入、缺失及替换编辑的t0代植株,对其进行基因型判定,共针对50株t0代植株进行判定,其中百分之4是嵌合体基因型,百分之20是纯合的双等位基因突变基因型,百分之30是双等位基因突变基因型,百分之26是杂合体基因型,百分之20是野生型基因型。
附图说明
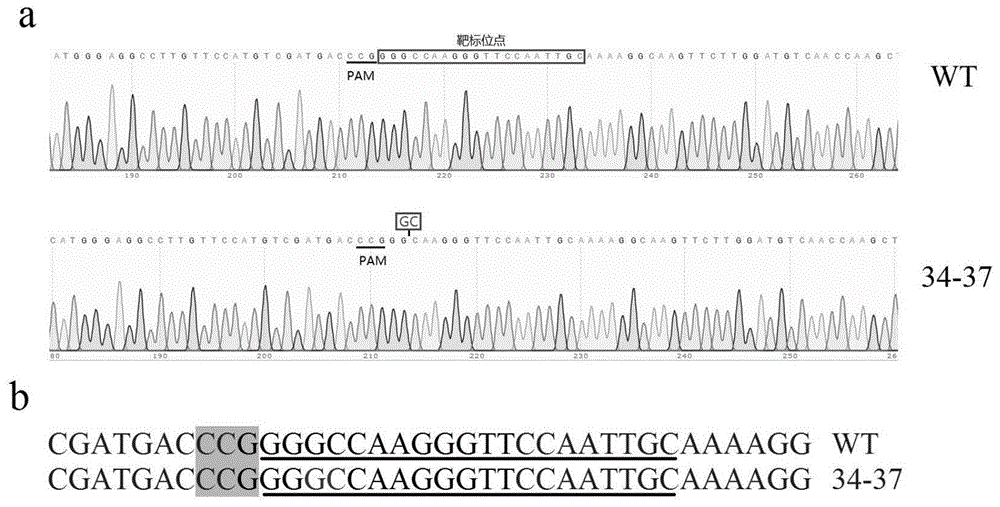
图1为双等位基因纯合体34-37测序图和序列,对应基因为oscao1,其中a为野生型wt和34-37的测序峰图,紫框圈住的序列为靶标序列,红框圈住的序列为缺失序列;b为野生型wt和34-37的部分序列,绿色标注pam,下划线标注靶标序列,红色标注缺失碱基;
图2为嵌合体植株34-44的测序图和序列,对应基因为oscao1,其中a为wt、34-44和34-44的t/a克隆34-44-1、34-44-2、34-44-3的测序峰图,紫框圈住的序列为靶标序列,蓝框圈住的序列为碱基替换,红框圈住的序列为缺失序列,红色倒三角指示测序峰图中出现两个及以上信号峰;b为wt、34-44-1、34-44-2和34-44-3的部分序列,绿色标注pam,下划线标注靶标序列,蓝色标注替换碱基,红色标注缺失碱基;
图3为双等位基因突变体34-33的测序图和序列,对应基因为oscao1,其中a为wt、34-33和34-33pcr产物t/a克隆34-33-1、34-33-2的测序峰图,紫框圈住的序列为靶标序列,绿框圈住的序列为插入序列,红框圈住的序列为缺失序列,红色倒三角指示测序峰图中出现两个及以上信号峰;b为wt、34-33、34-33-1和34-33-2的部分序列,绿色标注pam,下划线标注靶标序列,绿色标注插入碱基,红色标注缺失碱基;
图4为杂合体35-15测序图,对应基因为ossp1,其中a为wt、35-15和35-15pcr产物t/a克隆35-15-2、35-15-4的测序峰图,紫框圈住的的序列为靶标序列,红框圈住的的序列为缺失序列,红色倒三角指示测序峰图中出现两个及以上信号峰;b为wt、35-15-2和35-15-4的部分序列,绿色标注pam,下划线标注靶标序列,红色标注缺失碱基。
具体实施方式
本发明提供了一种快速鉴定二倍体基因编辑作物t0代基因型的方法,包括以下步骤:(1)以二倍体基因编辑作物t0代的基因组dna为模板dna,在基因编辑靶标位点的上下游设计特异性引物,进行pcr扩增,将得到的pcr产物回收后纯化和第一次sanger测序;当测序测序结果为单一峰图,则所述基因编辑作物t0代的植株为纯合植株;当测序结果单一峰后面出现3个信号峰,则为嵌合体;单一峰后面出现2个信号峰,则为杂合体或双等位基因突变基因型;
(2)对于测序结果有杂峰或套峰的所述pcr产物进行t/a克隆,得连接产物;
(3)将所述连接产物转化感受态细胞,在lb培养基中震荡培养60min后,将菌液涂抹在含氨苄青霉素的lb平板上,培养8~12h,挑取单菌落于含氨苄青霉素的lb培养基中震荡培养8h后,进行第二次sanger测序;将测序结果用ncbiblast工具,与野生型序列进行比对;基因编辑序列为三个不同的序列,则为嵌合体;基因编辑序列为两个不同的序列,则为双等位基因突变基因型;对应的两个序列,一个为野生型,一个为突变序列,则为杂合体基因型。
本发明以二倍体基因编辑作物t0代的基因组dna为模板dna,在基因编辑靶标位点的上下游设计特异性引物,进行pcr扩增,将得到的pcr产物回收后纯化和第一次sanger测序;当测序测序结果为单一峰图,则所述基因编辑作物t0代的植株为纯合植株;当测序结果单一峰后面出现3个信号峰,则为嵌合体;单一峰后面出现2个信号峰,则为杂合体或双等位基因突变基因型。本发明对所述基因编辑作物t0代的基因组dna提取方法并没有特殊限定,优选采用ctab法提取叶片的基因组dna。本发明优选将所述ctab法提取得到的dna沉淀溶解于30μl灭菌的双蒸水(含rnasea),37℃保温30min消化rna,4℃保存,然后再进行后续反应。
本发明优选在基因编辑靶标位点的下游350~500bp处设计特异性引物,更优选在所述基因编辑靶标位点的下游400bp处设计特异性引物。本发明所述特异性引物的tm值优选为58℃左右。在本发明中,利用所述特异性引物和阴性植株dna进行pcr扩增,pcr产物在1%琼脂糖,电压1.5v/cm条件下进行电泳,电泳条带单一,回收和纯化pcr产物后进行第一次sanger测序,测序结果没有杂峰。本发明对所述回收和纯化pcr产物的方法并没有特殊限定,利用本领域的常规pcr产物回收和纯化试剂盒即可。
本发明利用所述模板dna和特异性引物进行pcr扩增,所述pcr扩增的体系以20μl计,优选包括:模板dna1μl,10mmdntps0.5μl,所述特异性引物的上下游引物各0.5μl,10×extaqbuffer2μl,extaq0.1μl,加水补足至20μl;所述pcr扩增的反应程序优选包括:94℃预变性2min;94℃变性30s,56℃退火50s,72℃延伸40s,35个循环;72℃延伸5min。本发明所述模板dna的用量优选为40ng,所述extaq优选为购自takara的extaq(5u/μl,货号:rr001q);且所述pcr反应体系中特异性引物的上下游引物的浓度优选均为10μm。
本发明优选对所述pcr扩增得到的pcr产物按照universaldna纯化回收试剂盒(dp214)上的说明书进行纯化,纯化后的dna稀释到10ng/μl,取出10μl进行第一次sanger测序,如果测序结果仅为单一峰图,判定所述基因编辑作物t0代的植株为纯合植株,包括双等位基因纯合突变或阴性植株后,还包括用ncbiblast与野生型靶位点序列进行比对,来判断纯合基因型,如果和野生型序列一致,则为纯合阴性植株;如果不一致,则为双等位基因纯合突变;当上述pcr产物的测序结果为杂峰或套峰,根据单一峰后面出现的信号峰的个数,判定具体杂合基因型:单一峰后面出现3个信号峰,则所述基因编辑作物t0代的植株为嵌合体;单一峰后面出现2个信号峰,则所述基因编辑作物t0代的植株为杂合体或双等位基因突变基因型。
为了验证对嵌合体基因型的判断及区分杂合体或双等位基因突变基因型,本发明对于测序结果有杂峰或套峰的所述pcr产物进行t/a克隆,得连接产物。本发明所述t/a克隆优选利用cv21-zerobackgroundptopo-ta/bluntcloningkit进行,所述t/a克隆的体系以10μl计,优选包括:ptopo-bluntsimplevector1μl,pcr产物1μl,10×enhancer1μl和灭菌水7μl。本发明优选将所述体系在微量离心管中配制完成,混匀后25℃反应5min,完成t/a克隆。
得连接产物后,本发明将将所述连接产物转化感受态细胞,在lb培养基中震荡培养60min后,将菌液涂抹在含氨苄青霉素的lb平板上,培养8~12h,挑取单菌落于含氨苄青霉素的lb培养基中震荡培养8h后,进行第二次sanger测序;将测序结果用ncbiblast工具,与野生型序列进行比对;基因编辑序列为三个不同的序列,则为嵌合体;基因编辑序列为两个不同的序列,则为双等位基因突变基因型;对应的两个序列,一个为野生型,一个为突变序列,则为杂合体基因型。
本发明所述转化时,连接产物与感受态细胞的体积比优选为1:10,所述感受态细胞优选为jm109感受态细胞。本发明所述转化优选为将所述连接产物和感受态细胞混匀后,冰中静止25min,42℃热激45s,再在冰中放置3min,完成所述转化。本发明所述含氨苄青霉素的lb平板和含氨苄青霉素的lb培养基中氨苄青霉素的浓度优选均为100μg/ml。本发明所述震荡培养和培养的温度优选均为37℃。本发明通过降低连接产物的浓度,使最终得到的单菌落基本上都是单一基因型,将测序结果用ncbiblast工具进行比对,统计所得到的基因型。同时结合t0代植株测序结果判断植株可能基因型,即如果测序是单一峰则为纯合体,如果单一峰后面出现3个信号峰,则为嵌合体,如果单一峰后面出现2个信号峰,则可能为杂合体或双等位基因突变基因型。通过t载体测序结果和t0代植株测序结果来最终得到植株的基因型。
下面结合实施例对本发明提供的一种快速鉴定基因编辑作物t0代基因型的方法进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
试剂选择:taq酶为takara的extaq(货号:rr001q)
实验植株:转基因受体材料为粳稻品种日本晴,筛选标记为潮霉素。根据loc_os11g12740(ossp1)与loc_os10g41780(oscao1)的基因序列在网站http://crispr.hzau.edu.cn/cgi-bin/crispr2/crispr进行靶位点预测,每个基因挑选1个合适的crispr靶位点,ossp1所选靶位点在基因5’到3’方向对应序列为gccatggtgtcaccaccctt(seqidno.1),oscao1所选靶位点在基因5’到3’方向对应序列为gggccaagggttccaattgc(seqidno.2),根据靶位点设计靶位点接头引物。接头引物合成后,采用goldengate策略构建crispr载体,方法参见(maetal.,2015)。
实施例1
1、基因组dna提取
将新鲜叶片放入经液氮预冷的研钵中,研磨,尽量细。取少许材料加入1.5ml离心管中,加入600μl65℃预热的ctab提取液,快速混匀,65℃保温30min,不时颠倒混匀,待冷至室温后加入500μl氯仿/异戊醇(v1:v2=24:1),颠倒混匀使溶液呈乳浊状(不要振荡),以12,000r/min的转速,离心5min,取上清液,加入600μl预冷的异丙醇,颠倒混匀,以12,000r/min的转速,离心10min,沉淀dna,弃去上清液,依次用800μl70%(v/v)乙醇洗涤沉淀两次,稍干燥,将dna沉淀溶解于30μl灭菌的双蒸水(含rnasea),37℃保温30min消化rna,4℃保存。
2、靶标基因的pcr
在oscao1基因靶标位点gggccaagggttccaattgc上游239bp处设计引物f239:ccacaggcttatctctgacact(seqidno.3),tm=57℃,下游132bp处设计引物r132:tgtccaatgcttcctaccttgt(seqidno.4),tm=59℃;在ossp1基因靶标位点gccatggtgtcaccaccctt上游214bp处设计引物f214:ccaagaagttgcctgaccacac(seqidno.5),tm=60℃,下游237bp处设计引物r237:catcatctcgaacgcctggatc(seqidno.6),tm=59℃;
pcr反应体系:稀释的基因组dna1μl(约40ng),10mmdntps0.5μl,上下游引物(10μm)各0.5μl,10×extaqbuffer(mg2+plus)(20mm)2μl,takaraextaq(5u/μl)0.1μl,加水补足至20μl。反应程序:94℃,2min;94℃,30s,56℃,50s,72℃,40s,35cycles;72℃,5min,4℃保存。
pcr产物按照universaldna纯化回收试剂盒(dp214)上的说明书进行纯化,纯化后的dna稀释到10ng/μl,取出10μl进行测序。
3、纯合基因型判断
如果测序结果只为单一的峰图,如图1所示,则为双等位基因纯合突变或阴性植株,用ncbiblast与野生型靶位点序列进行比对,如果和野生型序列一致,则为阴性植株;如果不一致,则为双等位基因纯合突变。
4、杂合基因型
如果测序结果显示杂峰或套峰等,则可能为杂合体、嵌合体、双等位基因突变等杂合基因型,根据单一峰后面出现的信号峰的个数,判定具体基因型:单一峰后面出现3个信号峰,则所述基因编辑作物t0代的植株为嵌合体;单一峰后面出现2个信号峰,则所述基因编辑作物t0代的植株为杂合体或双等位基因突变基因型。将其pcr产物用于下面的克隆实验。
5、pcr产物克隆测序实验
选择cv21-zerobackgroundptopo-ta/bluntcloningkit进行t/a克隆实验,在微量离心管中配制下列体系(10μl体系,表1)。
表1t/a克隆体系
混匀后25℃反应5min,将全部连接产物(10μl)加入至100μljm109感受态细胞中,混匀,冰中静止25min,42℃热激45s后,再在冰中放置3min。加入500μllb培养基,37℃振荡培养60min。取200μl菌液涂到含有100μg/ml氨苄青霉素的lb平板上,37℃培养过夜,挑取10-20个白色菌落到含有1ml100μg/ml氨苄青霉素的lb液体培养基中,37℃振荡培养8h后,取200μl进行测序。
6、嵌合体基因型的验证及杂合体或双等位基因突变基因型的确定
通过降低连接产物的浓度,得到的单菌落基本上都是单一基因型,将测序结果用ncbiblast工具与野生序列进行比对,确定具体基因编辑序列,验证根据t0代植株测序结果判断植株杂合体基因型的准确性:即如果单一峰后面出现3个峰,如图2中的t0,则为嵌合体,对应的基因编辑序列为三个不同的序列;如果单一峰后面出现两个信号峰,对应的两个序列,一个为野生型,一个为突变序列,则为杂合体基因型,如图4中的t0;如果单一峰后面出现两个信号峰,对应的两个序列为不同的序列,则为双等位基因突变基因型,如图3中的t0。通过t载体测序结果和t0代植株测序结果来最终得到植株的基因型。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
序列表
<110>中国农业科学院油料作物研究所
<120>一种快速鉴定二倍体基因编辑作物t<sub>0</sub>代基因型的方法
<160>6
<170>siposequencelisting1.0
<210>1
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>1
gccatggtgtcaccaccctt20
<210>2
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>2
gggccaagggttccaattgc20
<210>3
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>3
ccacaggcttatctctgacact22
<210>4
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>4
tgtccaatgcttcctaccttgt22
<210>5
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>5
ccaagaagttgcctgaccacac22
<210>6
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>6
catcatctcgaacgcctggatc22
- 还没有人留言评论。精彩留言会获得点赞!