一种苯磺顺阿曲库铵的制备方法及其应用与流程
本发明属于药物合成领域,具体涉及一种苯磺顺阿曲库铵的制备方法及其应用,尤其涉及一种毒性小的苯磺顺阿曲库铵的制备方法及其应用。
背景技术:
苯磺酸阿曲库铵是一种非去极化肌肉松弛药,具有起效快、作用时间短,治疗剂量不影响心、肝、肾功能和无积蓄性等特点,临床广泛用于防治各种需要肌肉松弛或控制呼吸的情况。苯磺顺阿曲库铵是苯环酸阿曲库铵单一异构体,是新一代的肌松剂,作用强度约为阿曲库铵的3倍,通过非肝非肾代谢且不释放胺,具有心血管保护性。
cn107056699a公开了一种高纯度顺苯磺酸阿曲库铵的制备方法,包括以下步骤:化合物iii与苯磺酸反应,所得化合物iv发生取代反应生成化合物v,化合物v再与1,5-二氯戊烷反应生成顺苯磺酸阿曲库铵,该发明制备苯磺顺阿曲库铵时采用中间体与氢氧化钠或碳酸氢钠成钠盐,这样相当于将中间体提纯,无论是实验室中还是生产上反应条件容易满足和控制,生产效率高,副反应小,产品纯度高,且氢氧化钠或碳酸氢钠容易获得,价格相对便宜。但其过早甲基化导致过早出现手性异构体,降低了总收率。
现有技术中通常采用以下方法合成苯磺顺阿曲库铵:
但该方法存在以下问题:反应原料戊二醇二丙烯酸酯的制备比较复杂,需要高温精馏设备,一般普通生产车间无法生产;该工艺第一步的迈克尔加成反应中,生产放大有易爆的潜在风险;甲基化过程中会产生两个异构体,导致甲基化的反应收率不到60%。且该异构体去除需要硅胶柱层析纯化,溶剂消耗很大,产生大量固废硅胶;以及最后一步甲基化过程用到了苯磺酸甲酯,该试剂为基因毒性杂质,成品中残留很容易超标。因此,如何提供一种安全、异构体少、收率高、杂质低的苯磺顺阿曲库铵的制备方法,成为了亟待解决的问题。
技术实现要素:
针对现有技术的不足,本发明的目的在于提供一种苯磺顺阿曲库铵的制备方法及其应用,尤其提供一种毒性小的苯磺顺阿曲库铵的制备方法及其应用。本发明提供的制备方法生产过程安全,收率高,异构体少,最大单杂低。
为达到此发明目的,本发明采用以下技术方案:
一方面,本发明提供了一种苯磺顺阿曲库铵的制备方法,所述制备方法包括以下步骤:
(1)将r-四氢罂粟碱-n-乙酰-l-亮氨酸盐溶解,用碱性溶液调ph值,之后与3-溴丙酸甲酯和碱混合,反应,得到中间体a;
(2)将步骤(1)得到的中间体a与碱混合,反应,得到中间体b;
(3)将步骤(2)得到的中间体b与甲基化试剂混合,反应,得到中间体c;
(4)将步骤(3)得到的中间体c、催化剂和脱水剂混合,反应,得到所述苯磺顺阿曲库铵。
反应式如式i所示:
本发明通过季铵化反应替代迈克尔加成反应,提高了生产安全;步骤(3)中间体c的制备过程(甲基化过程)异构体少,并且可通过过滤方式去除,无需柱层析,成本大大降低,更加环保;甲基化过程被提前,反应过程中甲基化试剂残留更易控制;总体收率高,成本低,最大单杂低至0.1%以下,杂质种类少,且在生产过程中可控,成品药物更安全。
优选地,步骤(1)所述碱性溶液的溶质包括有机碱和/或无机碱,所述有机碱包括三乙胺、二异丙基乙胺、二乙胺或dbu(1,8-二氮杂二环十一碳-7-烯)中任意一种或至少两种的组合,例如三乙胺和二乙胺的组合、三乙胺和dbu的组合或二异丙基乙胺和二乙胺的组合等,所述无机碱包括氢氧化钾、氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、碳酸铯或氨水中任意一种或至少两种的组合,例如氢氧化钾和氢氧化钠的组合、碳酸钠和碳酸铯的组合或氢氧化锂和碳酸钾的组合等,但不限于以上所列举的组合,上述组合范围内其他未列举的组合同样适用,优选碳酸钠。
优选地,步骤(1)所述碱包括有机碱和/或无机碱,所述有机碱包括三乙胺、二异丙基乙胺、二乙胺或dbu中任意一种或至少两种的组合,例如三乙胺和二乙胺的组合、三乙胺和dbu的组合或二异丙基乙胺和二乙胺的组合等,所述无机碱包括氢氧化钾、氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、碳酸铯或氨水中任意一种或至少两种的组合,例如氢氧化钾和氢氧化钠的组合、碳酸钠和碳酸铯的组合或氢氧化锂和碳酸钾的组合等,但不限于以上所列举的组合,上述组合范围内其他未列举的组合同样适用。
优选地,步骤(1)所述r-四氢罂粟碱-n-乙酰-l-亮氨酸盐与3-溴丙酸甲酯的摩尔比为1:1-1:1.4。
优选地,步骤(1)所述r-四氢罂粟碱-n-乙酰-l-亮氨酸盐与碱的摩尔比为1:1-1:1.4。
优选地,步骤(1)所述反应的温度为40-90℃,优选70-80℃。
优选地,步骤(1)所述反应的时间为20-28h。
优选地,步骤(1)所述ph值为8-10。
其中r-四氢罂粟碱-n-乙酰-l-亮氨酸盐与3-溴丙酸甲酯的摩尔比可以是1:1、1:1.1、1:1.2、1:1.3或1:1.4等,r-四氢罂粟碱-n-乙酰-l-亮氨酸盐与碱的摩尔比可以是1:1、1:1.1、1:1.2、1:1.3或1:1.4等,反应的温度可以是40℃、50℃、60℃、70℃、80℃或90℃等,时间可以是20h、21h、22h、23h、24h、25h、26h、27h或28h等,ph值可以是8、8.5、9、9.5或10等,但不限于以上所列举的数值,上述数值范围内其他未列举的数值同样适用。
优选地,步骤(2)所述碱包括有机碱和/或无机碱,所述有机碱包括三乙胺、二异丙基乙胺、二乙胺或dbu中任意一种或至少两种的组合,例如三乙胺和二乙胺的组合、三乙胺和dbu的组合或二异丙基乙胺和二乙胺的组合等,所述无机碱包括氢氧化钾、氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、碳酸铯或氨水中任意一种或至少两种的组合,例如氢氧化钾和氢氧化钠的组合、碳酸钠和碳酸铯的组合或氢氧化锂和碳酸钾的组合等,但不限于以上所列举的组合,上述组合范围内其他未列举的组合同样适用,优选氢氧化钠。
优选地,步骤(2)所述中间体a与碱的摩尔比为1:3-1:6。
优选地,步骤(2)所述混合的温度为0-15℃,优选5-10℃。
优选地,步骤(2)所述反应的温度为20-30℃。
优选地,步骤(2)所述反应的时间为20-28h。
其中,中间体a与碱的摩尔比可以是1:3、1:4、1:5或1:6等,混合的温度可以是0℃、1℃、2℃、3℃、4℃、5℃、6℃、7℃、8℃、9℃、10℃、11℃、12℃、13℃、14℃或15℃等,反应的温度可以是20℃、21℃、22℃、23℃、24℃、25℃、26℃、27℃、28℃、29℃或30℃等,时间可以是20h、21h、22h、23h、24h、25h、26h、27h或28h等,但不限于以上所列举的数值,上述数值范围内其他未列举的数值同样适用。
优选地,步骤(3)所述中间体b与甲基化试剂的摩尔比为1:1.8-1:2.2。
优选地,步骤(3)所述甲基化试剂包括苯磺酸甲酯。
优选地,步骤(3)所述反应的温度为15-45℃,优选25-30℃。
优选地,步骤(3)所述反应的时间为20-28h。
其中,中间体b与甲基化试剂的摩尔比可以是1:1.8、1:1.9、1:2、1:2.1或1:2.2等,反应的温度可以是15℃、20℃、25℃、30℃、35℃、40℃或45℃等,时间可以是20h、21h、22h、23h、24h、25h、26h、27h或28h等,但不限于以上所列举的数值,上述数值范围内其他未列举的数值同样适用。
上述将中间体a水解为中间体b的步骤在先、中间体b甲基化为中间体c的步骤在后能够减少过早甲基化带来的异构体杂质含量大,进而提高了反应收率,减少了最大单杂,提高了成品安全性。
优选地,步骤(4)所述中间体c与催化剂的摩尔比为1:1.5-1:2.5。
优选地,步骤(4)所述中间体c与脱水剂的摩尔比为1:1.5-1:2.5。
优选地,步骤(4)所述催化剂包括苯磺酸、硫酸或盐酸中任意一种。
优选地,步骤(4)所述脱水剂包括无水硫酸钙、无水硫酸镁、无水硫酸钠或分子筛中任意一种。
优选地,步骤(4)所述反应的温度为15-55℃。
优选地,步骤(4)所述反应的时间为20-28h。
其中,中间体c与催化剂的摩尔比可以是1:1.5、1:1.6、1:1.7、1:1.8、1:1.9、1:2、1:2.1、1:2.2、1:2.3、1:2.4或1:2.5等,中间体c与脱水剂的摩尔比可以是1:1.5、1:1.6、1:1.7、1:1.8、1:1.9、1:2、1:2.1、1:2.2、1:2.3、1:2.4或1:2.5等,温度可以是15℃、20℃、25℃、30℃、35℃、40℃、45℃、50℃或55℃等,,时间可以是20h、21h、22h、23h、24h、25h、26h、27h或28h等,但不限于以上所列举的数值,上述数值范围内其他未列举的数值同样适用。
上述步骤(4)在中间体b甲基化为中间体c的步骤之后,能够减少甲基化过程中甲基化试剂残留至成品中,减少了最大单杂,提高了成品安全性。
作为本发明优选的技术方案,所述制备方法包括以下步骤:
(1)将r-四氢罂粟碱-n-乙酰-l-亮氨酸盐溶解,用碱性溶液调ph值至8-10,之后与3-溴丙酸甲酯和碱混合,在40-90℃下反应20-28h,得到中间体a;
(2)将步骤(1)得到的中间体a与碱在0-15℃下混合,之后在20-30℃下反应20-28h,得到中间体b;
(3)将步骤(2)得到的中间体b与甲基化试剂混合,在15-45℃下反应20-28h,得到中间体c;
(4)将步骤(3)得到的中间体c、催化剂和脱水剂混合,在15-55℃下反应20-28h,得到所述苯磺顺阿曲库铵。
另一方面,本发明还提供了所述的苯磺顺阿曲库铵的制备方法在制备非去极化肌肉松弛药中的应用。
与现有技术相比,本发明具有如下有益效果:
本发明通过优化制备方法流程提供了一种苯磺顺阿曲库铵的制备方法,所述制备方法利用季铵化反应替代迈克尔加成反应,提高了生产安全;步骤(3)中间体c的制备过程(甲基化过程)异构体少,并且可通过过滤方式去除,无需柱层析,成本大大降低,更加环保;甲基化过程被提前,反应过程中甲基化试剂残留更易控制;总体收率高,成本低,最大单杂低至0.1%以下,杂质种类少,且在生产过程中可控,成品药物更安全。
附图说明
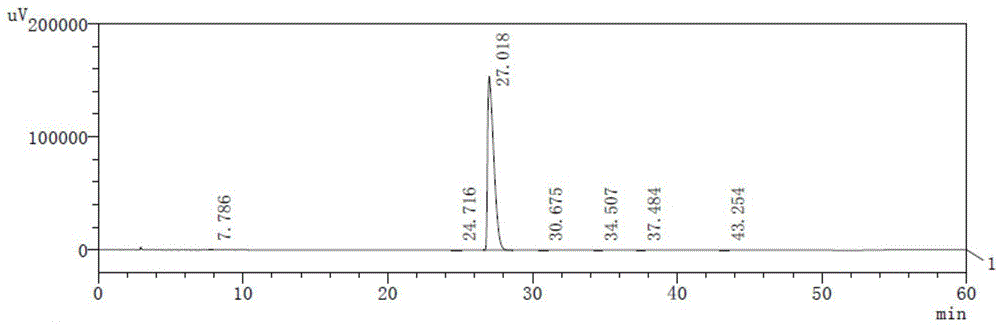
图1是实施例2得到的产品的hplc图;
图2是对比例1得到的产品的hplc图;
图3是对比例2得到的产品的hplc图。
具体实施方式
为更进一步阐述本发明所采取的技术手段及其效果,以下结合本发明的优选实施例来进一步说明本发明的技术方案,但本发明并非局限在实施例范围内。
以下实施例中所有原料均可从商业获得。
以下实施例和对比例中,顺苯纯度和最大单杂的检测方法为hplc,色谱柱名称:kromasil100-5c18,长度:250mm,内径:4.6mm,波长:280nm。
实施例1
本实施例提供了一种苯磺顺阿曲库铵的制备方法,反应式如下:
具体步骤如下:
(1)取r-四氢罂粟碱-n-乙酰-l-亮氨酸盐(50g,0.0967mol)在甲苯(250ml)和水(250ml)中用20%碳酸钠水溶液调ph为9,分液,有机层在55℃减压浓缩至不滴;然后加入乙腈(250ml)溶解后,再加入3-溴丙酸甲酯(19.4g,0.116mol)和无水碳酸钠(12.3g,0.116mol),在80℃下反应24h;反应完毕,冷却至20℃过滤;过滤液在40℃减压浓缩至不滴,加入丙酮(500ml)溶解,加入草酸(10.4g,0.116mol)成盐析晶,湿品烘干后得到中间体a(47.7g),收率:95.0%;
(2)取步骤(1)得到的中间体a(47.7g,0.0918mol)加入乙腈(238ml)和水(477ml),在5℃下滴加氢氧化钠的水溶液(氢氧化钠:14.7g,0.3675mol溶于147ml水中),滴加完毕,在25℃反应24h,反应完毕,用苯磺酸水溶液调ph为8,将水减压浓缩出,加入甲苯(100ml)带水两次,得中间体b(65g),收率:100%,无需纯化,直接进行下一步反应;
(3)取步骤(2)得到的中间体b(65g,0.0918mol)加入乙腈(238ml)和苯磺酸甲酯(31.6g,0.1836mol)在25℃下反应24h,冷却后过滤,滤饼减压干燥后得到中间体c(40.5g),收率:75.1%;
(4)将步骤(3)得到的中间体c(40.5g,0.0689mol)、苯磺酸(21.8g,0.1378mol)、无水硫酸钙(18.8g,0.1378mol)和二氯甲烷(405ml)混合,45℃回流分水反应24h,反应结束后,过滤不溶物,在5℃下,用苯磺酸水溶液(203ml,ph4)洗3次,二氯甲烷层干燥后在25℃减压浓缩至122ml,滴入乙醚(610ml)析晶。真空干燥后得到苯磺顺阿曲库铵成品(35.1g),收率:82.0%。顺苯纯度:99.3%,最大单杂:0.05%。四步反应总收率58.3%,表征数据如下:
中间体a结构表征:1hnmrδ(cdcl3):2.75-2.78(4h,m),3.00-3.07(1h,m),3.23-3.26(3h,m),3.38-3.41(1h,m),3.51(4h,s),3.65(3h,s),3.81(3h,s),3.84(3h,s),3.85(3h,s),4.15-4.17(1h,d)5.84(1h,s),6.56-6.64(3h,m),6.74-6.76(1h,d),8.61(2h,br).
esi+ms(m/z):430.8[m-1个草酸+1]+。
中间体b结构表征:1hnmrδ(d2o):2.35-2.48(4h,m),2.75-2.88(5h,m),2.98-3.00(1h,m),3.17(3h,s),3.50(3h,s),3.55(3h,s),3.57(3h,s),3.82-3.85(1h,m),5.62(1h,s),6.25-6.27(2h,d),6.37(1h,s),6.52-6.54(1h,d).
esi+ms(m/z):416.3[m+1]+
中间体c结构表征:1hnmrδ(d2o):2.38-2.75(4h,m),2.80-2.87(5h,m),2.98-3.03(1h,m),3.40(3h,s),3.68(3h,s),3.81(3h,s),3.82(3h,s),3.83(3h,s),4.72-4.75(1h,m),5.81(1h,s),6.47-6.48(1h,d),6.63-6.65(1h,m),6.86(1h,s),6.89-6.91(1h,m),7.41-7.46(3h,m),7.83-7.85(2h,m).
esi+ms(m/z):430.6[m-1个苯磺酸根]+
苯磺顺阿曲库铵成品结构表征:1hnmrδ(cdcl3):1.54-1.56(2h,m),1.67-1.69(4h,m),2.83-2.87(2h,m),2.90-2.94(2h,m),3.12-3.17(2h,m),3.20(6h,s),3.26-3.31(2h,m),3.39(8h,s),3.51-3.58(4h,m),3.64(6h,s)3.79(12h,s),3.84-3.87(2h,m),4.01-4.05(2h,m),4.12-4.15(4h,m),4.19-4.22(2h,m),4.93-4.94(2h,m),5.93-4.94(2h,s),6.42-6.43(2h,d),6.47(2h,s),6.53(2h,s),6.63-6.65(2h,d),7.28-7.30(6h,m),7.82-7.83(4h,m).
esi+ms(m/z):464.6[(m-2个苯磺酸根)/2]2+。
实施例2
本实施例提供了一种苯磺顺阿曲库铵的制备方法,反应式如下:
具体步骤如下:
(1)取r-四氢罂粟碱-n-乙酰-l-亮氨酸盐(50g,0.0967mol)在甲苯(250ml)和水(250ml)中用三乙胺调ph为8,分液,有机层在55℃减压浓缩至不滴;然后加入乙腈(250ml)溶解后,再加入3-溴丙酸甲酯(16.2g,0.0967mol)和三乙胺(9.8g,0.0967mol),在40℃下反应28h;反应完毕,冷却至20℃过滤;过滤液在40℃减压浓缩至不滴,加入丙酮(500ml)溶解,加入草酸(10.4g,0.116mol)成盐析晶,湿品烘干后得到中间体a(47.1g),收率:93.86%;
(2)取步骤(1)得到的中间体a(47.1g,0.0906mol)加入乙腈(238ml)和水(477ml),在0℃下滴加二乙胺(24g,0.3288mol),滴加完毕,在20℃反应28h,反应完毕,用盐酸调ph为9,将水减压浓缩出,加入甲苯(100ml)带水两次,得中间体b(63.7g),收率100%,无需纯化,直接进行下一步反应;
(3)取步骤(2)得到的中间体b(63.7g,0.0906mol)加入乙腈(238ml)和苯磺酸甲酯(31.2g,0.1812mol)在15℃下反应28h,之后过滤,滤饼减压干燥后得到中间体c(40.1g);
(4)将步骤(3)得到的中间体c(40.1g,0.0682mol)、苯磺酸(16.19g,0.1024mol)、无水硫酸钠(14.5g,0.1024mol)和二氯甲烷(405ml)混合,40℃回流分水反应28h,反应结束后,过滤不溶物,在5℃下,用苯磺酸水溶液(203ml,ph4)洗3次,二氯甲烷层干燥后在25℃减压浓缩至122ml,滴入乙醚(610ml)析晶。真空干燥后得到苯磺顺阿曲库铵成品(34.7g),顺苯纯度:99.1%,最大单杂:0.09%,收率81.8%,四步反应总收率57.6%。
实施例3
本实施例提供了一种苯磺顺阿曲库铵的制备方法,反应式如下:
具体步骤如下:
(1)取r-四氢罂粟碱-n-乙酰-l-亮氨酸盐(50g,0.0967mol)在甲苯(250ml)和水(250ml)中用氢氧化钠水溶液调ph为10,分液,有机层在55℃减压浓缩至不滴;然后加入乙腈(250ml)溶解后,再加入3-溴丙酸甲酯(22.6g,0.1354mol)和氢氧化钠(5.4g,0.1354mol),在90℃下反应20h;反应完毕,冷却至20℃过滤;过滤液在40℃减压浓缩至不滴,加入丙酮(500ml)溶解,加入草酸(10.4g,0.116mol)成盐析晶,湿品烘干后得到中间体a(46.3g),收率:92.0%;
(2)取步骤(1)得到的中间体a(46.3g,0.0891mol)加入乙腈(238ml)和水(477ml),在0℃下滴加碳酸钾水溶液(碳酸钾:89.4g,0.6468mol溶于200ml水中),滴加完毕,在30℃反应20h,反应完毕,用苯磺酸调ph为8,将水减压浓缩出,加入甲苯(100ml)带水两次,得中间体b(62.1g),收率100%,无需纯化,直接进行下一步反应;
(3)取步骤(2)得到的中间体b(62.1g,0.0891mol)加入乙腈(238ml)和苯磺酸甲酯(40.8g,0.1782mol)在45℃下反应20h,之后过滤,滤饼减压干燥后得到中间体c(39.7g);
(4)将步骤(3)得到的中间体c(39.7g,0.0676mol)、苯磺酸(26.72g,0.1689mol)、无水硫酸镁(20.3g,0.1689mol)和二氯甲烷(405ml)混合,55℃回流分水反应20h,反应结束后,过滤不溶物,在5℃下,用苯磺酸水溶液(203ml,ph4)洗3次,二氯甲烷层干燥后在25℃减压浓缩至122ml,滴入乙醚(610ml)析晶。真空干燥后得到苯磺顺阿曲库铵成品(34.2g),顺苯纯度:99.2%,最大单杂:0.1%,四步反应总收率56.8%。
对比例1
本对比例提供了一种苯磺顺阿曲库铵的制备方法,反应式如下:
制备方法参照专利wo9200965(a1)如下:
(1)取r-四氢罂粟碱-n-乙酰-l-亮氨酸盐(50g,0.0967mol)在甲苯(250ml)和水(250ml)中用氢氧化钠水溶液调ph为10,分液,有机层在55℃减压浓缩至不滴;然后加入甲苯(250ml)溶解后,再加入戊二醇二丙烯酸酯(10.3g,0.04835mol)和醋酸(2.5ml),在70℃下反应4h;反应完毕,冷却至20℃过滤;过滤液在40℃减压浓缩至不滴,加入丙酮(500ml)溶解,加入草酸(10.4g,0.116mol)成盐析晶,湿品烘干后得到中间体1(44.4g),收率:85.1%;
(2)取步骤(1)得到的中间体1(44.4g,0.0411mol)溶于水(1.2l)中,用碳酸钠(质量分数20%的水溶液)调ph为7.0-8.0,加入甲苯(600ml)萃取,有机层在50-60℃之间减压浓缩后黄色粘稠液体,加入苯磺酸甲酯(70ml),25℃反应过夜,加入甲苯和水,分层后水相冷冻干燥得到黄色固体,黄色固体溶于二氯甲烷中,进行柱层析纯化,用混合溶剂(v(二氯甲烷):v(甲醇):v(苯磺酸)=80:20:0.5)进行洗脱,收集馏分,用氯化钠溶液(质量分数10%)洗涤一次,有机层在25℃减压浓缩干。用水(150ml)溶解,并用苯磺酸调ph为4.0,冷冻干燥后得到苯磺顺阿曲库铵成品(18.0g),顺苯纯度:98.2%,最大单杂:0.3%,两步反应总收率为29.9%。
对比例2
本对比例提供了一种苯磺顺阿曲库铵的制备方法,反应式如下:
制备方法参照专利wo2008132746(a1)如下:
(1)取r-四氢罂粟碱-n-乙酰-l-亮氨酸盐(50g,0.0967mol)在水(200ml)中,用质量分数25%的氨水溶液调ph为9-10之间,加入二氯甲烷(200ml)在25℃下搅拌15min,分液,水相再用二氯甲烷(3×200ml)萃取,合并有机层,用质量分数10%氯化钠溶液(200ml)洗涤一次,用无水硫酸镁(40g)干燥,有机层在25℃减压浓缩至不滴;然后加入苯(100ml)溶解后,再加入丙烯酸甲酯(20g,0.2326mol)和醋酸(2.6ml),在80℃下反应4h;反应完毕,冷却至20℃过滤;加入丙酮(500ml)溶解,加入草酸(18.4g,0.146mol)的丙酮(90ml)溶液,然后加入乙酸乙酯(200ml),过滤,湿品在50℃烘干后得到中间体1(45.0g),收率:89.5%;
(2)取步骤(1)得到的中间体1(45.0g,0.0866mol)溶于水中(550ml),用质量分数25%的氢氧化钠溶液调ph为9-10之间,加入甲苯(275ml)在25℃下搅拌15min,分液,水层再用甲苯(2×275ml)萃取,合并有机层用质量分数10%氯化钠水溶液(100ml)萃取,用无水硫酸镁(20g)干燥后减压浓缩,加入乙腈(27.5ml)和苯磺酸甲酯(36.4g,0.2116mol)在30℃反应20h,加入二氯甲烷(83ml)搅拌成均相,加入乙醚(110ml)搅拌12h,过滤,湿品用50ml乙醚-二氯甲烷混合溶液(v:v=4:3)漂洗后,将过滤液浓缩干,加入乙酸乙酯(80ml)和乙醚(210ml)搅拌2h,过滤,湿品用50ml乙酸乙酯-乙醚混合溶液(v:v=3:8)漂洗,干燥后得到中间体2(25.3g),收率:48.5%
(3)将步骤(2)得到的中间体2(25.3g,0.0420mol)加入的水(100ml),苯磺酸(1.33g,0.0084mol)在40℃搅拌10h,反应完毕,减压浓缩至不滴,加入甲苯(30ml)带水1次,得到中间体3(27.0g)收率:100%。
(4)将步骤(3)得到的中间体3(27.0g,0.0420mol)溶于无水二氯甲烷(540ml)中,降温至0℃,在该温度下滴加草酰氯(6.3g,0.050mol),滴加完毕后恢复至25℃搅拌2h,随后温度降至0℃,滴加1,5-戊二醇(2.18g,0.021mol),滴加完毕恢复至25℃搅拌4h,在25℃下减压浓缩至不滴,加入水(500ml)和甲苯(1000ml)混合液,分液,水相用混合溶剂(v(乙酸乙酯):v(正庚烷)=5:1,1000ml)洗涤一次,接着用甲苯(1000ml)洗涤一次,再用二氯甲烷(200ml×2)将物料萃取出,干燥后减压浓缩得到苯磺顺阿曲库铵(12.4g)纯度:98.2%,最大单杂:0.5%。收率:47.5%,四步反应总收率为20.6%。
总结以上实施例对比例可以看出,本发明提供的制备方法通过优化制备方法流程,具有收率高、最大单杂在0.1%以下、杂质种类少、质量可控性好、产品安全性高的效果。
之后对实施例2和对比例1-2得到的产品进行hplc检测(色谱柱为kromasil100-5c18,规格为250mm×4.6mm×5μm),结果分别见图1-3。从图中可以发现,本发明提供的方法得到的产品相比对比例1-2,杂质种类数量出现明显降低,说明本发明提供的制备方法得到的产品质量可控性好、安全性更高。
申请人声明,本发明通过上述实施例来说明本发明的苯磺顺阿曲库铵的制备方法及其应用,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
- 还没有人留言评论。精彩留言会获得点赞!