一种糖基和多肽修饰的1,2,3-三唑化合物的制备方法
本发明涉及一种糖基和多肽修饰的1,2,3-三唑化合物的制备方法,属于有机合成与化学生物学相结合的技术领域。
背景技术:
1,2,3-三唑化合物是一类重要的五元含氮杂环结构骨架,在生物化学以及药物化学中有着广泛的应用。对于三唑骨架的修饰已成为制取医药、农药及杀虫剂的重要方法。例如向三唑母体结构中引入糖基和多肽对其生物活性、环境适应性及药理学性质均会产生显著的影响。目前已有的合成该类化合物的方法很少,且底物范围窄,反应条件苛刻并且大多需要过渡金属的参与。因此需要提供一种条件温和、简单高效且适用范围广的糖基和多肽修饰的1,2,3-三唑化合物的合成方法。
技术实现要素:
本发明的目的是提供一种糖基和多肽修饰的1,2,3-三唑化合物的制备方法,该制备方法简单,操作方便,成本较低,收率高。
本发明提供的糖基和多肽修饰的1,2,3-三唑化合物的制备方法,包括如下步骤1)或2):
1)在三乙烯二胺的催化下,式1或式1’所示化合物、式2所示化合物与式3所示化合物或肽进行反应即得式ⅰ或式ⅰ’所示1,2,3-三唑化合物;
2)在三乙烯二胺的催化下,式1或式1’所示化合物、式2所示化合物与肽进行反应即得式ⅱ或式ⅱ’所示1,2,3-三唑化合物;
式1或式1’中,x选自氟、氯和溴,y选自碳、氮、氧和硫;
式1’、式ⅰ’和式ⅱ’中,y’选自c6~10的芳基,优选苯基或卤素取代的苯基,最优选4-溴苯基;
式ⅰ、式ⅰ’和式1中,基团a为来自于糖的基团;
式2中,r1选自金属离子或硅基,所述金属离子优选为钠离子,所述硅基优选为c1~c3烷基取代的硅基;
式ⅰ、式ⅰ’和式3中,r2为取代或未经取代的烷基;
式ⅱ和式ⅱ’中,基团r3为来自于所述肽的基团。
上述的制备方法中,所述反应的溶剂可为四氢呋喃、乙腈、1,2-二氯乙烷或乙酸乙酯。
上述的制备方法中,所述反应在空气气氛中进行;
所述反应的温度可为25~80℃,时间可为5~48h。
上述的制备方法中,式1或式1’所示化合物、式2所示化合物、式3所示化合物、所述肽与所述三乙烯二胺的摩尔比可为1:1~4:1~4:1~4:0.5~2,如1:4:3:3:2。
上述的制备方法中,所述糖可为单糖、二糖或多糖;
所述单糖可为葡萄糖、果糖、半乳糖、核糖、脱氧核糖或蔗糖;
优选地,式1所示化合物为三氟甲基取代丙酮叉保护的果糖、三氟甲基取代丙酮叉保护的葡萄糖或三氟甲基取代丙酮叉保护的2’-脱氧尿苷;
式1’所示化合物为三氟甲基取代的对溴苯乙酮。
上述的制备方法中,所述肽可为二肽、三肽或多肽;
所述二肽可为双甘肽、蛋甘肽、苯丙甘肽或谷胱甘肽。
上述的制备方法中,r2为未取代或取代的环烷基;
优选地,所述环烷基的碳原子数为5~7;
所述环烷基上的取代基为酯基、羰基或羟甲基;
式3所示化合物优选为脯氨酸甲酯或四氢吡咯。
上述的制备方法中,式ⅰ所示糖基和多肽修饰的1,2,3-三唑化合物为下述式ⅰ-1-式1-4所示化合物中任一种:
式ⅱ所示糖基和多肽修饰的1,2,3-三唑化合物为式ⅱ-1所示化合物:
式ⅱ’所示糖基和多肽修饰的1,2,3-三唑化合物为式ⅱ’-1所示化合物:
本发明提供的糖基和多肽修饰的1,2,3-三唑化合物的制备方法,克服了现有方法存在的底物范围窄、合成步骤繁琐、反应条件苛刻以及过渡金属催化剂参与等缺陷,该方法原料廉价易得,操作简单,不需无水无氧操作,在空气条件下即可顺利进行,在合成化学以及化学生物学领域均具有很高的价值。本发明方法制备的糖基和多肽取代的1,2,3-三唑表现出生物多样性,在荧光蛋白成像,活体检测和医药合成等方面具有很高的潜在应用价值。
附图说明
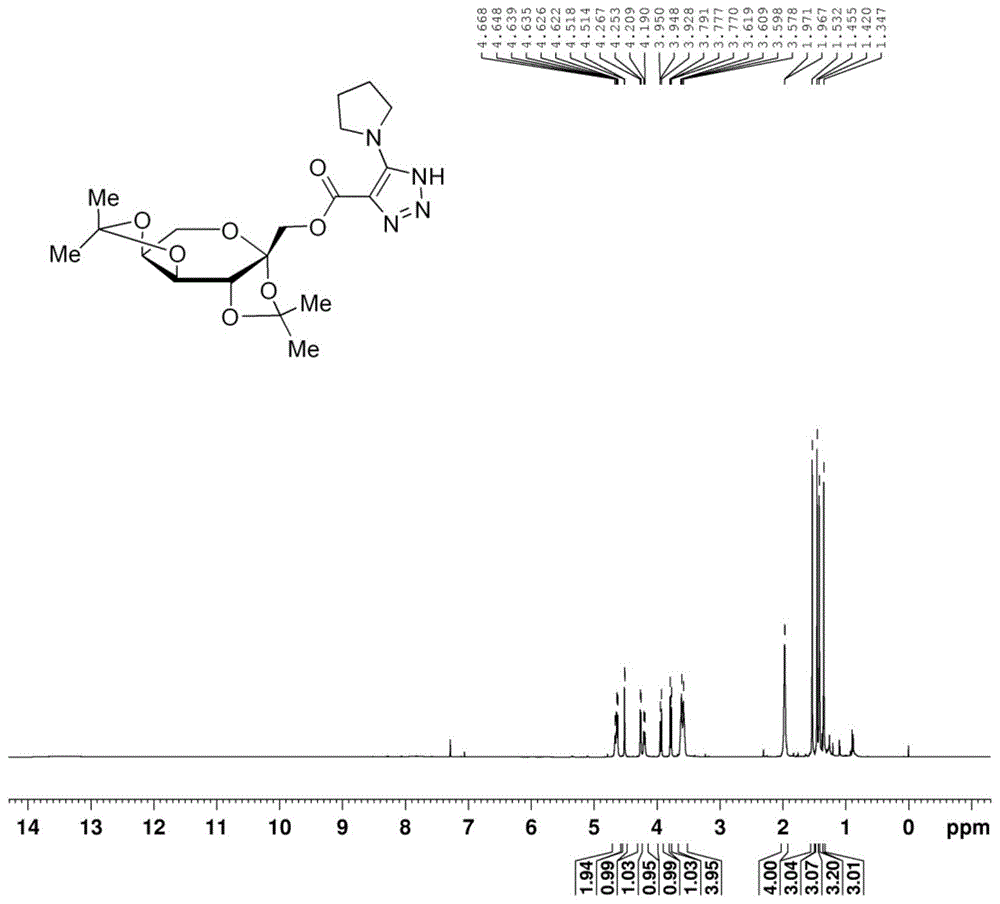
图1为式ⅰ-1所示((3ar,5as,8as,8br)-2,2,7,7-四甲基四氢-3ah-双([1,3]二氧戊环)[4,5-b:4',5'-d]吡喃-3a-基)甲基5-(吡咯烷-1-基)-1h-1,2,3-三唑-4-羧酸酯的核磁共振氢谱图。
图2为式ⅰ-2所示(3ar,5r)-5-((r)-2,2-二甲基-1,3-二氧戊环-4-基)-2,2-二甲基四氢呋喃基[2,3-d][1,3]二氧-6-基5-(吡咯烷-1-基)-1h-1,2,3-三唑-4-羧酸酯核磁共振氢谱图。
图3为式ⅰ-3所示(3ar,5r)-5-((r)-2,2-二甲基-1,3二氧戊环-4-基)-2,2-二甲基四氢呋喃基5-((s)-2-(甲氧基羰基)吡咯烷-1-基)-1h-1,2,3-三唑-4-羧酸酯核磁共振氢谱图。
图4为式ⅰ-4所示((3ar,4r,6r,6ar)-6-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-2,2-二甲基四氢呋喃基[3,4-d][1,3]二氧戊环-4-基)甲基5-(吡咯烷-1-基)-1h-1,2,3-三唑-4-羧酸酯核磁共振氢谱图。
图5为式ⅱ-1所示(3ar,5r)-5-((r)-2,2-二甲基-1,3-二氧戊环-4-基)-2,2-二甲基四氢呋喃基[2,3-d][1,3]二氧戊环-6-基5-((2-((2-乙氧基-2-氧乙基)氨基)-2-氧乙基)(甲基)氨基)-1h-1,2,3-三唑-4-羧酸酯核磁共振氢谱图。
图6为式ⅱ’-1所示乙基n-(4-(4-溴苯甲酰基)-1h-1,2,3-三唑-5-基)-n甲基甘氨酰基蛋氨酸盐核磁共振氢谱图。
具体实施方式
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1、式ⅰ-1所示化合物的制备
在空气条件下,向反应管中依次加入三氟甲基取代丙酮叉保护的果糖(式1)(0.2mmol)、dabco(0.4mmol)、nan3(0.8mmol)、四氢吡咯(式3)(0.6mmol)和四氢呋喃(1.0ml)。其自由升温至50℃,反应12小时。反应完毕后,加入20ml乙酸乙酯和3.0ml0.5m稀盐酸萃取分液,有机相再用3.0ml饱和食盐水洗涤,后干燥,旋转蒸发,柱层析。经称重,分离得到46mg式ⅰ-1所示化合物,产率为54%。
图1为本实施例制备的式ⅰ-1(((3ar,5as,8as,8br)-2,2,7,7-四甲基四氢-3ah-双([1,3]二氧戊环)[4,5-b:4',5'-d]吡喃-3a-基)甲基5-(吡咯烷-1-基)-1h-1,2,3-三唑-4-羧酸酯)的核磁共振氢谱图,表征如下:
1hnmr(600mhz,cdcl3)δ4.66(d,j=11.6hz,1h),4.63(dd,j=8.0,2.4hz,1h),4.52(d,j=2.4hz,1h),4.26(d,j=8.0hz,1h),4.20(d,j=11.6hz,1h),3.94(dd,j=12.8,1.2hz,1h),3.78(d,j=8.0hz,1h),3.62-3.58(m,4h),1.99-1.94(m,4h),1.53(s,3h),1.46(s,3h),1.42(s,3h),1.35(s,3h).
实施例2、式ⅰ-2所示化合物的制备
在空气条件下,向反应管中依次加入三氟甲基取代丙酮叉保护的葡萄糖(式1)(0.2mmol)、dabco(0.4mmol)、tmsn3(0.8mmol)、四氢吡咯(式3)(0.6mmol)和四氢呋喃(1.0ml)。其自由升温至50℃,反应12小时。反应完毕后,加入20ml乙酸乙酯和3.0ml0.5m稀盐酸萃取分液,有机相再用3.0ml饱和食盐水洗涤,后干燥,旋转蒸发,柱层析。经称重,分离得到52mg式ⅰ-2所示化合物,产率为61%。
图2为本发明实施例制备的(3ar,5r)-5-((r)-2,2-二甲基-1,3-二氧戊环-4-基)-2,2-二甲基四氢呋喃基[2,3-d][1,3]二氧-6-基5-(吡咯烷-1-基)-1h-1,2,3-三唑-4-羧酸酯核磁共振氢谱图,表征如下:
1hnmr(600mhz,cdcl3)δ13.6(br,0.8h),5.89(d,j=3.4hz,1h),5.45(s,1h),4.62(d,j=3.4hz,1h),4.31(s,2h),4.07(br,2h),3.56(br,4h),1.98(br,4h),1.53(s,3h),1.41(s,3h),1.31(s,3h),1.26(s,3h).
实施例3、式ⅰ-3所示化合物的制备
在空气条件下,向反应管中依次加入三氟甲基取代丙酮叉保护的葡萄糖(式1)(0.2mmol)、dabco(0.4mmol)、nan3(0.8mmol),脯氨酸甲酯(式3)(0.6mmol)和四氢呋喃(1.0ml)。其自由升温至80℃,反应12小时。反应完毕后,加入20ml乙酸乙酯和3.0ml0.5m稀盐酸萃取分液,有机相再用3.0ml饱和食盐水洗涤,后干燥,旋转蒸发,柱层析。经称重,分离得到58mg式ⅰ-3所示化合物,产率60%。
图3为本实施例制备的(3ar,5r)-5-((r)-2,2-二甲基-1,3二氧戊环-4-基)-2,2-二甲基四氢呋喃基5-((s)-2-(甲氧基羰基)吡咯烷-1-基)-1h-1,2,3-三唑-4-羧酸酯核磁共振氢谱图,表征如下:
1hnmr(600mhz,cdcl3)δ12.6(0.9h),5.96(d,j=3.7hz,1h),5.42(d,j=2.1hz,1h),4.83(d,j=5.2hz,1h),4.63(d,j=3.7hz,1h),4.37-4.34(m,2h),4.14-4.11(m,1h),4.08-4.06(m,1h),3.83-3.78(m,1h),3.71(s,3h),3.67-3.63(m,1h),2.35-2.28(m,1h),2.16-2.11(m,1h),2.05-1.99(m,2h),1.54(s,3h),1.42(s,3h),1.32(s,3h),1.29(s,3h).
实施例4、式ⅰ-4所示化合物的制备
在空气条件下,向反应管中依次加入三氟甲基取代丙酮叉保护的2’-脱氧尿苷(式1)(0.2mmol)、dabco(0.4mmol)、msn3(0.8mmol)、四氢吡咯(式3)(0.6mmol)和四氢呋喃(1.0ml)。其自由升温至50℃,反应12小时。反应完毕后,加入20ml乙酸乙酯和3.0ml0.5m稀盐酸萃取分液,有机相再用3.0ml饱和食盐水洗涤,后干燥,旋转蒸发,柱层析。经称重,分离得到46mg式ⅰ-4所示化合物,产率51%。
图4为本实施例制备的((3ar,4r,6r,6ar)-6-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-2,2-二甲基四氢呋喃基[3,4-d][1,3]二氧戊环-4-基)甲基5-(吡咯烷-1-基)-1h-1,2,3-三唑-4-羧酸酯核磁共振氢谱图,表征如下:
1hnmr(600mhz,cdcl3)δ10.1(br,1h),7.87(s,1h),5.94(s,1h),5.73(d,j=8.0hz,1h),5.17(d,j=4.6hz,1h),4.91(d,j=4.6hz,1h),4.61(s,1h),4.49(d,j=11.0hz,1h),4.45(d,j=11.0hz,1h),3.55(t,j=6.2hz,4h),1.95(t,j=6.2hz,4h),1.59(s,3h),1.35(s,3h).
实施例5、式ⅱ-1所示化合物的制备
在空气条件下,向反应管中依次加入三氟甲基取代丙酮叉保护的葡萄糖(式1)(0.2mmol)、dabco(0.4mmol)、nan3(0.8mmol)、双甘肽(0.6mmol)和四氢呋喃(1.0ml)。其自由升温至50℃,反应48小时。反应完毕后,加入20ml乙酸乙酯和3.0ml0.5m稀盐酸萃取分液,有机相再用3.0ml饱和食盐水洗涤,后干燥,旋转蒸发,柱层析。经称重,分离得到43mg式ⅱ-1所示化合物,产率41%。
图5为本实施例制备的(3ar,5r)-5-((r)-2,2-二甲基-1,3-二氧戊环-4-基)-2,2-二甲基四氢呋喃基[2,3-d][1,3]二氧戊环-6-基5-((2-((2-乙氧基-2-氧乙基)氨基)-2-氧乙基)(甲基)氨基)-1h-1,2,3-三唑-4-羧酸酯核磁共振氢谱图,表征如下:
1hnmr(600mhz,dmso-d6)δ13.2(br,1h),7.35(t,j=5.4hz,1h),5.97(d,j=3.6hz,1h),5.48(d,j=3.6hz,1h),4.66(d,j=3.7hz,1h),4.39-4.33(m,2h),4.22(q,j=7.2hz,2h),4.15-4.12(m,3h),4.10-4.01(m,3h),3.06(s,3h),1.54(s,3h),1.43(s,3h),1.32(s,3h),1.29(s,3h),1.28(t,j=7.2hz,3h).
实施例6、式ⅱ’-1所示化合物的制备
在空气条件下,向反应管中依次加入三氟甲基取代的对溴苯乙酮(0.2mmol)、dabco(0.4mmol)、tmsn3(0.8mmol)、蛋甘肽(0.6mmol)和四氢呋喃(1.0ml)。其自由升温至50℃,反应48小时。反应完毕后,加入20ml乙酸乙酯和3.0ml0.5m稀盐酸萃取分液,有机相再用3.0ml饱和食盐水洗涤,后干燥,旋转蒸发,柱层析。经称重,分离得到68mg式ⅱ’-1所示化合物,产率72%。
图6为本实施例制备的乙基n-(4-(4-溴苯甲酰基)-1h-1,2,3-三唑-5-基)-n甲基甘氨酰基蛋氨酸盐核磁共振氢谱图,表征如下:
1hnmr(600mhz,cdcl3)δ13.7(br,0.8h),7.97(d,j=8.2hz,2h),7.76(d,j=8.4hz,1h),7.56(d,j=8.2hz,2h),4.86-4.82(m,1h),4.23-4.20(m,2h),4.08(d,j=16.7hz,1h),3.91(d,j=16.7hz,1h),3.02(s,3h),2.67-2.59(m,2h),2.29-2.23(m,1h),2.12-2.07(m,1h),2.07(s,3h),1.27(t,j=7.2hz,3h).
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!