一种未知融合基因的检测方法与流程
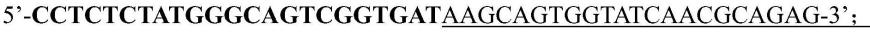
1.本发明属于基因检测技术领域,具体涉及一种未知融合基因的检测方法。
背景技术:
2.融合基因是指两个基因的全部或一部分的序列相互融合为一个新的基因的过程,其有可能是染色体易位、中间缺失或染色体倒置所致的结果。目前主要检测方法包括荧光原位杂交(fish)、逆转录聚合酶链反应(rt-pcr)、免疫组化(ihc)和高通量测序(ngs)等几种方法,从dna、rna和蛋白质等水平进行检测,这几种检测方法优劣并存,几种检测方法优缺点如下所述:
3.fish法,特异性高,结果形象直观,但是fish操作方法复杂、耗时长;不能够确认参与融合的基因,也不能够区分不同的融合类型,可能会错失发生率低或者较为复杂的重排形式的融合基因;常规fish方法需要计算不少于50个细胞,所以对于组织量少的标本(例如穿刺活检样本),fish不能操作;必须知晓发生融合的两个基因的序列来设计探针;两个发生融合的基因所在染色体的位置必须相距较近;不能同时检测不同基因与一个已知基因的融合。
4.rt-pcr法需根据不同融合方式设计不同的引物来进行检测,可操作性强、灵敏度高,但是只能检测单个融合基因,不能够识别新的融合基因伴侣或者解析复杂的结构重组,同时较为昂贵,导致该方法融合基因检测困难及检出率的降低。
5.ihc法虽然简单、便捷,但是其无法直接检测融合基因,对结果的判读主观性较强。
6.高通量测序的方法,虽然实验操作和数据分析方面很耗时,价格高,但是在现有的检测方法中,其准确率、灵敏度、特异性最高,一次可以检测多种基因和多种融合变体,可以发现未知变异。
技术实现要素:
7.本发明所要解决的技术问题是如何检测未知融合基因或如何提高检测未知融合基因的准确性。
8.为了解决上述技术问题,本发明首先提供了一种构建未知融合基因检测文库的方法。所述未知融合基因为未知基因与已知基因融合而形成的融合基因。所述未知融合基因含有目标基因,所述目标基因为序列已知的dna片段。所述构建方法包括如下步骤:
9.a1、将样本的总rna或片段化后的总rna加入逆转录体系中进行反应获得逆转录产物;所述逆转录体系含有逆转录酶、随机引物和tso-n引物;所述tso-n引物从5’到3’依次包含端头序列、通用序列1和3’末端3个鸟苷酸;所述逆转录酶具有模板转换和末端转移酶的活性,会在新合成的cdna的3’末端添加3个c;所述随机引物的序列为6-9个随机碱基;
10.a2、将a1所得逆转录产物加入第一轮扩增体系中进行pcr扩增,获得第一轮pcr扩增产物;所述第一轮扩增体系含有tso-f引物和第一特异性引物;所述tso-f引物从5’到3’依次包括接头序列1和通用序列1;所述第一特异性引物针对融合基因3’端区域设计;
11.a3、将第一轮pcr扩增产物放入第二轮扩增体系中进行扩增,获得未知融合基因检测文库;所述第二轮扩增体系含有所述tso-f引物、第二特异性引物和barcode引物;所述第二特异性引物为ω引物,从5’到3’依次包括5'端特异序列和通用序列2和3'端特异序列,所述5'端特异序列和3'端特异序列针对融合基因3’端区域设计,所述通用序列2不能与所述融合基因结合;所述barcode引物从5’到3’依次含有接头序列2、barcode特异序列和通用序列2;所述barcode特异序列用以区分不同样本,在不同样本之间序列不同。
12.所述barcode特异序列均为长度为6-12nt、无3个以上连续碱基。
13.所述tso-n引物的核苷酸序列为序列表中序列1所示,其中,所述通用序列1为序列表中序列1的第13-39位,所述3个鸟苷酸为序列表中序列1的第40-42位。
14.所述第二特异性引物的5'端特异序列和3'端特异序列针对所述第一特异性引物结合位置的上游区段设计。
15.上述方法中,所述a1步骤中的随机引物的序列可为5
′‑n6-9-3
′
,所述n
6-9
为6-9个n,所述n为a、g、c或t。所述随机引物的序列可为5
′‑n6-3
′
,所述n6为6个n,所述n为a、g、c或t。
16.所述构建未知融合基因检测文库的方法也可为包括如下步骤的方法:
17.b1、将样本的总rna在逆转录体系中进行逆转录或将所述总rna片段化后在逆转录体系中进行逆转录获得逆转录产物;所述逆转录体系含有逆转录酶、随机引物和tso-n引物;所述tso-n引物包含通用序列1和3’末端3个鸟苷酸;所述逆转录酶具有模板转换和末端转移酶的活性,会在新合成的cdna的3’末端添加3个c;所述随机引物的序列为5
′‑n6-9-3
′
,所述n
6-9
为6-9个n,所述n为a、g、c或t。
18.所述tso-n引物是核苷酸序列为序列表中序列1所示的单链dna,序列表中序列1的第13-39位为通用序列1,第40-42位为3个鸟苷酸。
19.序列表中序列1的第13-42位为tso(template switching oligo)序列。tso序列在5’端带有1个通用引物序列,而在3’末端,有3个核糖鸟苷(rg),可以促进模板转换,进而扩增出完整的cdna序列。
20.b2、进行第一轮pcr扩增:将b1所得产物在第一轮扩增体系中进行pcr扩增,获得第一轮pcr扩增产物;所述第一轮扩增体系含有tso-f引物和第一特异性引物;所述tso-f引物包括名称为接头序列1的用于测序的接头和所述通用序列1;所述第一特异性引物根据目标基因进行设计,引物能与所述目标基因特异结合。
21.所述第一轮pcr扩增产物是双链dna,一端是所述tso-f引物,另一端是所述第一特异性引物。
22.b3、第二轮pcr扩增,包括将a2所得产物在第二轮扩增体系中进行扩增,获得未知融合基因检测文库;所述第二轮扩增体系含有所述tso-f引物、第二特异性引物和barcode引物;所述第二特异性引物为ω引物,包括5'端特异序列和通用序列2和3'端特异序列,所述5'端特异序列和3'端特异序列根据所述目标基因进行设计,分别与所述目标基因的两个区段特异结合,所述通用序列2是位于所述5'端特异序列和所述3'端特异序列之间且不能与所述目标基因结合的一段单链dna;所述barcode引物用以区分不同样本和进行测序用,所述barcode引物含有名称为接头序列2的用于测序的接头、barcode特异序列和所述通用序列2;所述barcode特异序列用以区分不同样本,在不同样本之间序列不同。
23.上文所述随机引物的序列可为5
′‑n6-9-3
′
,所述n
6-9
为6-9个n,所述n为a、g、c或t。
所述随机引物的序列可为5
′‑n6-3
′
,所述n6为6个n,所述n为a、g、c或t。
24.上述b3步骤中所述第二特异性引物的5'端特异序列和3'端特异序列针对所述第一特异性引物结合位置的下游区段设计。
25.上述b3步骤中第二轮pcr扩增包含两个阶段,所述两个阶段的第一阶段为所述ts0-f引物和第二特异性引物组成引物对以所述第一轮pcr扩增产物为模板进行扩增,得到第一阶段的扩增产物;所述两个阶段的第二阶段为所述ts0-f引物和barcode引物组成引物对以所述第一阶段的扩增产物为模板进行扩增得到文库。
26.上文所述样本可为:新鲜组织、血液、石蜡包埋组织。
27.上述目标基因可为一种或多种。
28.所述接头序列1和所述接头序列2为测序接头,根据测序平台进行选择,用于将待测序列连接到测序平台的载体或芯片上并进行测序。
29.所述测序平台为illumina、mgi或iontorrent平台。
30.上文所述接头序列1和所述接头序列2为下述a或b的接头:
31.a、所述接头序列1与ion torrent测序平台的ion torrent接头p互补,所述接头序列2与ion torrent测序平台的芯片上的ion torrent接头a互补;
32.b、所述接头序列1与illumina测序平台的flowcell(流动池)上的p5接头互补,所述接头序列2与illumina测序平台的flowcell(流动池)上的p7接头互补。
33.如果测序平台为ion torrent平台,所述接头序列2与ion torrent平台测序引物互补,用来测序;所述接头序列1与平台载体上序列互补,用来将测序模板与平台载体连接;
34.如果测序平台为ion torrent平台,所述接头序列1和2分别为p和a。
35.如果测序平台为illumina平台,接头序列和芯片上的测序引物序列是互补的,并把核酸片段连接到载体上;
36.如果测序平台为illumina平台,所述接头序列1和2分别为i5和i7。
37.为了解决上述技术问题,本发明还提供了一种未知融合基因的检测方法,包括使用上文所述的构建方法构建未知融合基因检测文库,对所述未知融合基因检测文库进行测序,获得所述未知融合基因的序列。所述融合基因可为一种或多种。用于构建未知融合基因检测文库的产品也属于本发明的保护范围。所述产品含有上文所述tso-n引物、所述tso-f引物、所述第一特异性引物和第二特异性引物。
38.上文所述测序平台可为ion torrent平台,所述tso-f引物可为:所述tso-f引物含有接头序列1(加粗序列,针对ion torrent测序平台)和所述通用序列1(下划线标序列)。所述接头序列1根据测序平台iontorrent平台进行选择设计,为所述ion torrent测序平台的接头p。
39.上文所述产品还含有所述随机引物和/或所述barcode引物。
40.上文所述产品可为试剂/试剂盒或系统。
41.上述产品还可含有进行反转录和pcr扩增所需的其它试剂,如具有模板转换和末端转移酶活性的逆转录酶。
42.本文中,所述逆转录酶可为模板转换反转录酶(template switching rt enzyme)。
43.上文所述的构建方法、所述的检测方法或所述的产品在未知融合基因检测中的应用也属于本发明的保护范围。
44.本发明的方法检测样本类型为rna,使用特定逆转录酶在cdna的3'端加入3个c,然后tso-n序列3'端3个g与其互补,通过逆转录酶模板转换,再次以tso-n序列为模板完成延伸,最后所有反转录序列都加入一个共同的序列(即通用序列1),利用tso-f引物和第一特异性的目标基因引物进行第一轮扩增,以达到特异性扩增模板的作用,最后再经过一轮特异性扩增将需要检测的靶向区域进一步筛选以及标定特定样本的标签整合到构建好的文库中,再经过一次文库纯化步骤就得到用于上机测序的文库。通过生信分析可以调取与已知目标基因相融合的基因,以达到检测未知融合的目的,较传统的fish,免疫组化,qpcr,ddpcr以及ngs的常规检测方式具有了很大进步。
45.该方法所建文库根据不同的接头组合,可适用于当前主流的ngs测序平台,illumina、mgi以及iontorrent平台等。
46.创新及优势:
47.本发明提供了一种适用于低rna样本量的、且在只知晓发生融合的其中一个基因、而融合方式未知的情况下,能准确、特异性地检测融合基因的方法,能弥补现有技术的不足。具体为:
48.1.利用特定反转录酶在3’端加tso(template switching oligo)序列的特性,为cdna增加了固定末端序列,下一轮扩增即可实现特定基因的融合partner捕获;
49.2.rna水平的靶向未知融合检测,相较于dna水平的未知融合检测,得到的融合信息更准确,更真实,更直观;
50.3.相较于传统的未知融合检测,建库流程更简单,大大节约了检测和测序成本;
51.4.易于增减靶向检测的范围,panel的设置更灵活;
52.5.起始rna样本量要求低,检测成功率高;
53.6.基于二代测序平台,靶向未知融合的检测具有高灵敏度
54.7.两轮特异性引物的扩增,提高了目标片段的特异性,提高检测的准确性。
附图说明
55.图1为建库流程示意图。
56.图2为文库构建原理图。
57.图3为所构建未知融合检测文库2200质检结果。
具体实施方式
58.下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。以下提供的实施例可作为本技术领域普通技术人员进行进一步改进的指南,并不以任何方式构成对本发明的限制。
59.下述实施例中的实验方法,如无特殊说明,均为常规方法,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
60.实施例1、构建融合基因检测文库检测肺癌相关融合基因
61.文库构建
62.根据图1和2所示的流程,进行文库构建。使用iontorrent的genetron s5平台进行引物设计及后续上机测序。
63.一、样本rna提取
64.核酸提取:利用rna提取试剂盒(rneasy ffpe kit(50),qiagen,73504),进行样本核酸提取。
65.核酸定量及质控:样本核酸的定量和质控实验,采用试剂盒qubit rna hs assay kits(thermo fisher scientific,q32852),进行检测。
66.样本为5个肺癌ffpe样本,提取后,实际提取rna浓度使用qubit荧光计测定如下,提取的rna总量在50ng以上。
67.样本类型和样本提取rna浓度及纯度如下:
68.样本肿瘤类型样本类型qubit浓度(ng/ul)洗脱体积(ul)s1肺癌ffpe10045s2肺癌ffpe78.645s3肺癌ffpe54.845s4肺癌ffpe42.845s5肺癌ffpe22.845
69.二、样本反转录
70.1.热处理:
71.吸取待检样本rna(30-100ng),补无核酸酶水到3.5ul,涡旋混匀,瞬离一下,放入pcr仪中按照表1程序进行热处理(如果样本浓度低,3.5ul不能满足实验,可将反转录体系按比例扩大至20ul)。
72.表1 rna热处理反应条件
73.温度时间80℃10min25℃3min
74.2.逆转录:
75.热处理后的rna按照表2用随机引物和dntp(neb n0447v)配制反转录体系1(冰上配制),其中随机引物由6个随机碱基组成,序列为5
′‑
nnnnnn-3
′
,所述n为a、g、c或t。
76.表2 反转录体系1配制
77.体系1组分体积热处理后的rna3.5ul随机引物(100um)1.5uldntp(10mm)1ul总体积6ul
78.配制后用枪头上下吹吸混匀(至少10次),瞬离后,冰上备用。
79.按照表3配制反转录体系2(冰上配制),其中tso-n引物(上海生工生物工程有限公司)序列如下:
(序列表中序列1)包括:端头序列(加粗序列)、通用序列1(下划线标序列,序列表中序列1的第13-39位)和含有三个核糖核酸g碱基(rg)的“ggg”序列(序列表中序列1的第40-42位);逆转录酶template switching rt enzyme具有模板转换和末端转移酶的活性,会在新合成的cdna的3’末端添加c碱基,形成“ccc”序列:
80.表3 反转录体系2配制
[0081][0082]
配制后用枪头上下吹吸混匀(至少10次),瞬离,将配制好的反转录体系1(6ul)加到反转录体系2(4ul)上方,用枪头上下吹吸混匀(至少10次),瞬时离心,按表4程序进行cdna合成反应。
[0083]
表4 cdna合成反应条件
[0084]
温度时间42℃90min85℃5min4℃hold
[0085]
经过逆转录合成的cdna片段,首先在逆转录酶template switching rt enzyme mix(neb,m0466l)的作用下在cdna的3’端添加了“ccc”序列,然后反转录体系中的tso-n引物在3’末端有3个核糖鸟苷(rg),可以促进模板转换,进而逆转录酶再继续以tso-n引物为模板在cdna的“ccc”3’末端进行反转录延伸(图2),形成cdna的延伸序列(图2中cdna的“ccc”序列3’末端的片段)。
[0086]
3.纯化
[0087]
1)cdna合成结束后,加入18μl(1:1.6)ampure beads磁珠,涡旋混匀,室温静置10min;
[0088]
2)磁力架吸附至液体澄清,约5min后,弃上清;
[0089]
3)分别加入200μl现配的80%乙醇,孵育30s,弃上清;
[0090]
4)再次加入200μl现配80%乙醇,孵育30s,弃上清,除尽乙醇,磁力架上晾3min,待乙醇挥发完全;
[0091]
5)分别加入11μl水洗脱,涡旋混匀,室温静置10min;
[0092]
6)磁力架吸附至液体澄清,约5min后,吸取10μl上清至新的离心管,用于后续建库使用。
[0093]
三、第一轮pcr扩增
[0094]
1.第一轮特异性引物
[0095]
将纯化后的cdna全部用于第一轮pcr扩增,按照表5配制预扩增反应体系。其中的
ts0-f引物序列为:其含有接头序列1(加粗序列,本接头序列针对ion torrent测序平台)和通用序列1(下划线标序列),其中接头序列1根据测序平台iontorrent平台进行选择设计,为iontorrent测序平台的接头p;第一特异性引物(表6和图2)根据目标融合基因3’端区域设计,本发明中是根据6个常见相关基因ntrk1、alk、ros1、met、tbp、itgb7的容易发生融合的16个区域设计的,包括:f-ntrk1e10/f-ntrk1e11/f-ntrk1e12/f-ntrk1e13/f-ntrk1e17/f-ntrk1e7/f-ntrk1e8/f-ntrk1e9/f-alke19/f-alke20/f-rose32/f-rose34/f-rose35/f-mete15/f-itgb7/f-tbp其中的一条或一条以上,上述引物的序列见表6。
[0096]
表5 预扩增反应体系
[0097][0098]
表6 第一特异性引物和第二特异性引物列表
[0099]
[0100][0101]
注:“e”代表外显子,如“mete15”代表met基因的第15个外显子;第二特异性引物序列中5’端小写字体序列为5’端特异序列,3’端小写字体序列为3’端特异序列,中间大写字体序列为通用序列2“tctgtacggtgacaaggcg”。
[0102]
配制完成后盖紧pcr管盖,涡旋振荡混匀,进行短暂离心。按照表7设置预扩增程序:
[0103]
表7 预扩增反应条件
[0104][0105]
2.纯化
[0106]
1)预扩增结束后,加入39μl(1:1.3)ampure beads,涡旋混匀,室温静置10min;
[0107]
2)磁力架吸附至液体澄清,约5min后,弃上清;
[0108]
3)分别加入200μl现配的80%乙醇,孵育30s,弃上清;
[0109]
4)再次加入200μl现配80%乙醇,孵育30s,弃上清,除尽乙醇,磁力架上晾3min,待乙醇挥发完全;
[0110]
5)分别加入11μl水洗脱,涡旋混匀,室温静置10min;
[0111]
6)磁力架吸附至液体澄清,约5min后,吸取10μl上清至新的离心管,得到第一轮pcr扩增产物,用于后续建库使用。
[0112]
四、第二轮pcr,文库构建
[0113]
1.pcr扩增
[0114]
将第一轮pcr扩增产物全部用于文库构建,按照表8配制pcr扩增反应体系:
[0115]
表8 pcr扩增反应体系
[0116][0117]
第二轮pcr扩增引物包括tso-f引物、第二特异性引物和barcode引物,其中ts0-f引物与第一轮pcr扩增中相同。第二轮pcr扩增包含两个阶段,第一阶段为ts0-f引物和第二特异性引物组成引物对以第一轮的pcr扩增产物为模板进行扩增,进一步捕获目标基因;第二阶段为ts0-f引物和barcode引物组成引物对以第一阶段的扩增产物为模板进行扩增得到文库。其中第二特异性引物为ω引物,包括:5'端特异序列和通用序列2和3'端特异序列(表6中表注),其中的5'端特异引物和3'端特异引物同样是根据目标融合基因3’端区域设计的,本发明中也是根据步骤三中扩增中的6个基因的16个区域设计,但第二特异性引物的5'端特异序列和3'端特异序列针对第一特异性引物结合位置的上游区域设计(图2),包括:s-ntrk1e10/s-ntrk1e11/s-ntrk1e12/s-ntrk1e13/s-ntrk1e17/s-ntrk1e7/s-ntrk1e8/s-ntrk1e9/s-alke19/s-alke20/s-rose32/s-rose34/s-rose35/s-mete15/s-itgb7/s-tbp其中的一条或一条以上,序列见表6和图2。barcode引物包括接头序列2(本接头序列针对ion torrent测序平台)、barcode特异序列和通用序列2。barcode特异序列是与检测样本相对应的,不同样品之间的barcode特异序列不同;barcode特异序列均为长度为6-12nt、无3个以上连续碱基,本实施例中,barcode特异序列均为长度为11nt;接头序列2和通用序列2同样用于杂交捕获目标基因进行后续测序,接头序列2为ion torrent测序平台芯片上的ion torrent接头a。
[0118]
其中,barcode引物具体序列如表9所示:
[0119]
表9 barcode引物序列
[0120][0121]
配制完成后盖紧pcr管盖,涡旋振荡混匀,进行短暂离心。按照表10设置预扩增程序:
[0122]
表10 预扩增反应条件
[0123][0124]
2.纯化
[0125]
1)pcr扩增结束后,加入36μl(1:1.2)ampure beads,涡旋混匀,室温静置10min;
[0126]
2)磁力架吸附至液体澄清,约5min后,弃上清;
[0127]
3)分别加入200μl现配的80%乙醇,孵育30s,弃上清;
[0128]
4)再次加入200μl现配80%乙醇,孵育30s,弃上清,除尽乙醇,磁力架上晾3min,待乙醇挥发完全;
[0129]
5)分别加入32μl水洗脱,涡旋混匀,室温静置10min;
[0130]
6)磁力架吸附至液体澄清,约5min后,吸取30μl上清至新的离心管,得到构建的文库。
[0131]
构建文库后,用qubit 3.0荧光定量仪(invitrogen q33216)定量文库浓度。
[0132]
用aglient 2200生物分析仪检测文库条带大小分布如图3所示,文库浓度在1ng/ul以上和文库片段长度在200bp左右,构建的文库均匀,文库构建合格,可上机测序,每个样本需要200m数据量。
[0133]
五、高通量测序
[0134]
将上述构建的文库上机测序,使用iontorrent的genetron s5平台进行后续上机测序。
[0135]
融合测序下机分析数据的基本信息如表11所示:
[0136]
表11 测序下机分析数据的基本信息
[0137]
样本号s1s2s3s4s5total reads10668991306396144807515962351148883mapping rate66.72%67.70%65.45%60.18%65.09%tbp(对照)83681291810736185346856itgb7(对照)1969418675132391821983
[0138]
注:total reads代表测序得到的所有短序列或短片段的数目;mapping rate代表测序数据与参考基因组hg19的比对率;tbp为内参基因,用作样本正常的对照;itgb7为内参基因,用作样本正常的对照。
[0139]
六、生物信息分析:
[0140]
1.数据预处理:
[0141]
将原始bam文件转为fastq文件,并去除建库过程中引入的接头序列和低质量碱基片段。
[0142]
2.序列比对:
[0143]
首先,将fastq文件中的碱基序列比对到hg19(grch37)人类参考基因组上,生成
bam文件,然后进行排序;从排序的bam文件中获取候选融合序列后,再次比对到hg19上。
[0144]
3.融合分析:
[0145]
根据比对结果,分析融合的具体断点位置,并对融合断点进行注释。其中融合的检测及过滤参考文献[1][2]。
[0146]
其中,融合检测结果示例及说明如表12所示:
[0147]
表12 检测结果示例及说明
[0148][0149][0150]
参考文献:
[0151]
[1]huanying ge,kejun liu,todd juan,fang fang,matthew newman,wolfgang hoeck,fusionmap:detecting fusion genes from next-generation sequencing data at base-pair resolution,bioinformatics,volume 27,issue 14,15 july 2011,pages 1922
–
1928,https://doi.org/10.1093/bioinformatics/btr310.
[0152]
[2]d.nicorici,m.satalan,h.edgren,s.kangaspeska,a.murumagi,o.kallioniemi,s.virtanen,o.kilkku,fusioncatcher
–
a tool for finding somatic fusion genes in paired-end rna-sequencing data,biorxiv,nov.2014,doi:10.1101/011650。
[0153]
检测到的结果如下,其中将上述样本通过市售试剂盒:人类8基因突变联合检测试剂盒(半导体测序法)(北京泛生子基因科技有限公司)进行检测,然后结果表13所示,结果表明本发明的检测融合基因的方法检测到样本s1存在融合基因“eml4e20_alke20”,即eml4基因的20号外显子与目标基因alk的20号外显子存在融合(通过表6中的f-alke20和s-alke20引物检测得到);样本s2和s3均存在“met14skip”即met基因14外显子的缺失(通过表
6中的f-mete15和s-mete15引物检测得到);样本s4和s5未检测到融合基因。使用人类8基因突变联合检测试剂盒对这些样本的融合基因检测结果与上述检测结果一致,表明本发明所提供的检测方法可以有效检测待测样本的未知融合基因。
[0154]
表13 肺癌相关融合基因检测结果
[0155][0156]
注:“skip”代表缺失
[0157]
实施例2构建融合基因检测文库并检测肉瘤相关融合基因
[0158]
根据图1和2所示的流程,进行文库构建,使用iontorrent的genetron s5平台进行后续上机测序。
[0159]
样本信息如下。
[0160]
一、样本rna提取
[0161]
核酸提取:利用rna提取试剂盒paxgene blood rna kit-ivd(qiagen,762134),进行样本核酸提取。
[0162]
核酸定量及质控:样本核酸的定量和质控实验,采用试剂盒qubit rna hs assay kits(thermo fisher scientific,q32852),进行检测。
[0163]
样本为3个肉瘤骨髓组织样本(融合阴性),提取后,rna总量在50ng以上。
[0164]
具体如下:
[0165]
样本类型和样本提取rna浓度及纯度如下:
[0166]
样本肿瘤类型样本类型qubit浓度(ng/ul)洗脱体积(ul)1肉瘤骨髓142.1502肉瘤骨髓79.8503肉瘤骨髓72.350
[0167]
二、样本反转录
[0168]
1.rna打断处理:
[0169]
吸取待检rna(30-100ng),补无核酸酶水到2ul,涡旋混匀,瞬离一下,按照表14程序进行体系配制,然后涡旋混匀,瞬离一下,在pcr仪上94℃3.5min设置程序进行打断(温度升到94℃再放到pcr仪上),得到片段化的rna用于反转录。
[0170]
表14 rna打断反应体系
[0171]
试剂体积rna2ul2xfrag buffer1ul无核酸酶水1ul
[0172]
注:2xfrag buffer,stranded mrna-seqnlib prep module for illumina(rk20349)中包含的打断试剂。
[0173]
2.反转录:
[0174]
打断后的rna按照表15用template switching rt enzyme mix试剂盒(neb,m0466l)和dntp(neb,n0447v)配制反转录体系1(冰上配制):
[0175]
表15 反转录体系1配制
[0176]
体系1组分体积打断rna3.5ul随机引物(100um)1.5uldntp(10mm)1ul总体积6ul
[0177]
配制后用枪头上下吹吸混匀(至少10次),瞬离后,冰上备用,按照表16配制反转录体系2(冰上配制):
[0178]
表16 反转录体系2配制
[0179]
体系2组分体积template switching rt buffer2.5ultso-n(75um)0.5ultemplate switching rt enzyme mix1ul总体积4ul
[0180]
配制后用枪头上下吹吸混匀(至少10次),瞬离,将配制好的反转录体系1(6ul)加到反转录体系2(4ul)上方,用枪头上下吹吸混匀(至少10次),瞬时离心,按表17程序进行cdna合成反应。
[0181]
表17 cdna合成反应条件
[0182]
温度时间42℃90min85℃5min4℃hold
[0183]
3.纯化
[0184]
1)cdna合成结束后,加入18μl(1:1.6)ampure beads,涡旋混匀,室温静置10min;
[0185]
2)磁力架吸附至液体澄清,约5min后,弃上清;
[0186]
3)分别加入200μl现配的80%乙醇,孵育30s,弃上清;
[0187]
4)再次加入200μl现配80%乙醇,孵育30s,弃上清,除尽乙醇,磁力架上晾3min,待乙醇挥发完全;
[0188]
5)分别加入11μl水洗脱,涡旋混匀,室温静置10min;
[0189]
6)磁力架吸附至液体澄清,约5min后,吸取10μl上清至新的离心管,用于后续建库使用。
[0190]
三、第一轮pcr扩增
[0191]
1.第一轮pcr扩增
[0192]
将纯化后的cdna全部用于预扩增,按照表18配制预扩增反应体系:
[0193]
表18 预扩增反应体系
[0194][0195][0196]
配制完成后盖紧pcr管盖,涡旋振荡混匀,进行短暂离心。按照表19设置预扩增程序:
[0197]
表19 预扩增反应条件
[0198][0199]
2.纯化
[0200]
1)预扩增结束后,加入39μl(1:1.3)ampure beads,涡旋混匀,室温静置10min;
[0201]
2)磁力架吸附至液体澄清,约5min后,弃上清;
[0202]
3)分别加入200μl现配的80%乙醇,孵育30s,弃上清;
[0203]
4)再次加入200μl现配80%乙醇,孵育30s,弃上清,除尽乙醇,磁力架上晾3min,待乙醇挥发完全;
[0204]
5)分别加入11μl水洗脱,涡旋混匀,室温静置10min;
[0205]
6)磁力架吸附至液体澄清,约5min后,吸取10μl上清至新的离心管,用于后续建库使用。
[0206]
四、第二轮pcr扩增,文库构建
[0207]
1.pcr扩增
[0208]
将预扩增后的产物全部用于文库构建,按照表20配制pcr扩增反应体系:
[0209]
表20 pcr扩增反应体系
[0210][0211]
配制完成后盖紧pcr管盖,涡旋振荡混匀,进行短暂离心。按照表21设置预扩增程序:
[0212]
表21 预扩增反应条件
[0213][0214]
2.纯化
[0215]
1)pcr扩增结束后,加入36μl(1:1.2)ampure beads,涡旋混匀,室温静置10min;
[0216]
2)磁力架吸附至液体澄清,约5min后,弃上清;
[0217]
3)分别加入200μl现配的80%乙醇,孵育30s,弃上清;
[0218]
4)再次加入200μl现配80%乙醇,孵育30s,弃上清,除尽乙醇,磁力架上晾3min,待乙醇挥发完全;
[0219]
5)分别加入32μl水洗脱,涡旋混匀,室温静置10min;
[0220]
6)磁力架吸附至液体澄清,约5min后,吸取30μl上清至新的离心管。
[0221]
构建文库后,用qubit 3.0荧光定量仪定量文库浓度。用aglient 2200检测一下文库条带大小分布是否符合预期,文库合格后即可上机测序,每个样本需要200m数据量。
[0222]
融合测序下机数据的基本信息如下表所示:
[0223]
表22 测序下机数据基本信息
[0224]
样本123total reads254076932169942528023mapping rate0.53680.64540.5537tbp478729419itgb7438533814412
[0225]
生信分析过程如实施例1所述。
[0226]
检测到的融合形式如表23所示,结果表明三个检测样本均未检测到融合基因。
[0227]
表23 肉瘤样本检测结果
[0228][0229][0230]
以上对本发明进行了详述。对于本领域技术人员来说,在不脱离本发明的宗旨和范围,以及无需进行不必要的实验情况下,可在等同参数、浓度和条件下,在较宽范围内实施本发明。虽然本发明给出了特殊的实施例,应该理解为,可以对本发明作进一步的改进。总之,按本发明的原理,本技术欲包括任何变更、用途或对本发明的改进,包括脱离了本技术中已公开范围,而用本领域已知的常规技术进行的改变。按以下附带的权利要求的范围,可以进行一些基本特征的应用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1