一种目的基因多拷贝整合到酿酒酵母染色体rDNA的方法
to auxotrophic markers.biotechnol lett.35,1303
‑
7.
9.semkiv,m.v.,dmytruk,k.v.,sibirny,a.a.,2016.development of a system for multicopy gene integration in saccharomyces cerevisiae.j microbiol methods.120,44
‑
9.
10.shi,s.,liang,y.,zhang,m.m.,ang,e.l.,zhao,h.,2016.ahighly efficient single
‑
step,markerless strategy for multi
‑
copy chromosomal integration of large biochemical pathways in saccharomyces cerevisiae.metab eng.33,19
‑
27.
11.wang,l.,deng,a.,zhang,y.,liu,s.,liang,y.,bai,h.,cui,d.,qiu,q.,shang,x.,yang,z.,he,x.,wen,t.,2018.efficient crispr
‑
cas9 mediated multiplex genome editing in yeasts.biotechnol biofuels.11,277.
12.wei,s.,liu,y.,wu,m.,ma,t.,bai,x.,hou,j.,shen,y.,bao,x.,2018.disruption of the transcription factors thi2p and nrm1p alleviates the post
‑
glucose effect on xylose utilization in saccharomyces cerevisiae.biotechnol biofuels.11,112.
13.yang,x.,liu,j.,zhang,j.,shen,y.,qi,q.,bao,x.,hou,j.,2021.quorum sensing
‑
mediated protein degradation for dynamic metabolic pathway control in saccharomyces cerevisiae.metab eng.64,85
‑
94.
技术实现要素:
14.针对现有技术的不足,本发明要解决的问题是提供一种目的基因多拷贝整合到酿酒酵母染色体rdna的方法。
15.本发明所述目的基因多拷贝整合到酿酒酵母染色体rdna的方法,是将待转化的酿酒酵母先在含有羟基脲的培养基上进行预培养,然后再通过常规转化方法将两端带有rdna同源序列的dna片段转入酿酒酵母,最后在筛选培养基上筛选转化子实现;其特征在于:所述含有羟基脲的培养基是指在酵母常规培养基的基础上,添加终浓度为60mm
‑
250mm的羟基脲;所述在含有羟基脲的培养基上预培养的方法是:将待转化的酿酒酵母活化后,转接到含羟基脲的培养基中培养,按每两天转接一次,每次都是转接到新鲜的含有羟基脲的培养基中,总共培养6
‑
8天;所述两端带有rdna同源序列的dna片段含有筛选标记基因表达盒和目的基因表达盒,其组成及顺序为3
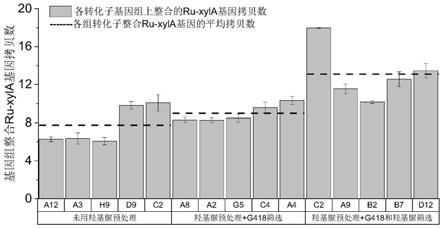
’‑
rdna同源序列1
‑
筛选标记基因表达框
‑
目的基因表达框
‑
rdna同源序列2
‑5’
;其中rdna同源序列1或rdna同源序列2是5s rdna或35s rdna区域的同源序列,筛选标记基因表达框或目的基因表达框上下游顺序随意,目的基因表达框可以是一个或多个;其中筛选标记基因是抗生素抗性基因或是弥补营养缺陷型酵母的营养缺陷的基因;所述筛选培养基针对的是引入转化子并表达的筛选标记基因,是带有抗生素的培养基或是营养缺陷型培养基;或是在所述筛选培养基基础上再额外添60mm
‑
250mm浓度的羟基脲。
16.上述目的基因多拷贝整合到酿酒酵母染色体rdna的方法中:所述含有羟基脲的培养基优选是指在酵母常规培养基的基础上,添加终浓度为150mm
‑
200mm的羟基脲。
17.上述目的基因多拷贝整合到酿酒酵母染色体rdna的方法中:所述rdna同源序列1或rdna同源序列2优选是35srdna区域的同源序列;所述的抗生素抗性基因优选是g418抗性
基因。
18.上述目的基因多拷贝整合到酿酒酵母染色体rdna的方法中:所述两端带有rdna同源序列的dna片段优选是rdna
up
‑
gfp
‑
kanmx
‑
ru
‑
xyla
‑
rdna
down
,其是利用seq id no:2所示核苷酸序列的引物rdna
‑1‑
f和seq id no:5所示核苷酸序列的引物rdna
‑2‑
r,以seq id no:1所示核苷酸序列的质粒peasy
‑
blunt
‑
gfp
‑
kanmx
‑
ruxi扩增获得。
19.上述目的基因多拷贝整合到酿酒酵母染色体rdna的方法中:所述筛选培养基优选是带有抗生素和60mm
‑
200mm羟基脲的培养基;或是营养缺陷型培养基加入终浓度150mm
‑
200mm羟基脲。
20.其中,进一步优选的实施方式是:所述筛选培养基是带有200mg/l
‑
20000mg/l g418,同时含有60mm
‑
200mm羟基脲的培养基;再进一步优选的实施方式是:所述筛选培养基是带有10000mg/l
‑
20000mg/l g418,同时含有60mm
‑
90mm羟基脲的培养基。最优选的实施方式是:所述筛选培养基是带有20000mg/l g418,同时含有60mm羟基脲的yepd培养基。
21.本发明公开的一种目的基因多拷贝整合到酿酒酵母染色体rdna的方法,是依据染色体上rdna是多拷贝重复单元,并且rdna拷贝数维持动态平衡的特点,先利用羟基脲刺激细胞,使部分rdna拷贝从染色体上丢失,之后将目的基因整合在剩余的rdna拷贝上。当去除环境中的羟基脲后,rdna区域按其拷贝数恢复机制出现不等姐妹染色单体重组。这一过程中,整合在rdna中的目的基因拷贝数随着rdna拷贝数的增加而增加。本发明首次利用rdna拷贝动态平衡的特点,建立了一次转化过程即在rdna上整合了18个拷贝目的基因的方法。该方法以rdna为整合靶点,操作便捷,无需额外引入除了重组dna片段之外的其他dna;并且,由于其所依赖的特殊机制,理论上可以和现有其他各种高拷贝重组方法联用,实现目的基因更高拷贝的整合,为基因编辑又提供了一种方法和有力的工具。
附图说明
22.图1:一种将目的基因多拷贝整合到酿酒酵母染色体rdna的方法操作流程示意图。
23.图2:质粒peasy
‑
blunt
‑
gfp
‑
kanmx
‑
ruxi结构。
24.图3:常规方法获得的转化子的相对荧光强度。
25.其中出发菌株直接用常规醋酸锂转化法转化,在含有20000mg/l g418的yepd培养基上筛选转化子,获得的转化子中随机挑选九十六个测定它们的相对荧光强度。
26.图4:羟基脲处理降低菌株染色体上rdna的拷贝数。
27.显示随着羟基脲处理时间的增加,菌株染色体上rdna的拷贝数逐渐降低。
28.图5:羟基脲预处理后再用常规方法获得的转化子的相对荧光强度。
29.其中出发菌株先用羟基脲预处理八天,再用常规醋酸锂转化法转化预处理后的菌株,在含有20000mg/l g418的yepd培养基上筛选转化子,在获得的转化子中随机挑选九十六个测定它们的相对荧光强度。
30.图6:羟基脲预处理后再用常规方法转化并在含羟基脲平板上筛选获得的转化子的相对荧光强度。
31.其中出发菌株先用羟基脲预处理八天,再用常规醋酸锂转化法转化预处理后的菌株,在含有20000mg/l g418以及60mm羟基脲的yepd培养基上筛选转化子,在获得的转化子中随机挑选九十六个测定它们的相对荧光强度。
32.图7:不同方法获得的转化子整合外源基因的拷贝数。
具体实施方式
33.下面结合具体附图和实施例对本发明内容进行详细说明。如下所述例子仅是本发明的较佳实施方式而已,应该说明的是,下述说明仅仅是为了解释本发明,并非对本发明作任何形式上的限制,凡是依据本发明的技术实质对实施方式所做的任何简单修改,等同变化与修饰,均属于本发明技术方案的范围内。
34.下述实施例中,所使用的材料、菌株、质粒、试剂等,如无特殊说明,均从商业途径得到。本发明涉及的方法均为遗传工程和分子生物学领域使用的常规技术和方法。例如methods in yeast genetics and genomics:a cold spring harbor laboratory course manual 2015 edition(cold spring harbor,n.y.:cold spring harbor laboratory press,2005)。
35.实施例1:转化用dna片段rdna
up
‑
gfp
‑
kanmx
‑
ru
‑
xyla
‑
rdna
down
的构建
36.本发明中,转化用dna片段组成如图1中所示,包括:rdna同源臂1、rdna同源臂2、多个基因表达盒。
37.在本实施例中,转化用的dna片段为rdna
up
‑
gfp
‑
kanmx
‑
ru
‑
xyla
‑
rdna
down
,是图2所示质粒peasy
‑
blunt
‑
gfp
‑
kanmx
‑
ruxi(seq id no:1)标注了fragment
‑
f和fragment
‑
r之间的那一部分。其具体组成包括:35srdna
‑
1(对应图1中的rdna同源臂1),木糖异构酶基因表达盒tef1p
‑
ru
‑
xyla
‑
adh1t,g418抗性基因表达盒tef1p
‑
kanmx
‑
tef1t,荧光报告基因表达盒tef1p
‑
yegfp
‑
pgkt,以及35srdna
‑
2(对应图1中的rdna同源臂2)。
38.其中,35s rdna
‑
1利用引物rdna
‑1‑
f(seq id no:2)和rdna
‑1‑
r(seq id no:3),以酵母染色体为模板扩增获得。35srdna
‑
2利用引物rdna
‑2‑
f(seq id no:4)和rdna
‑2‑
r(seq id no:5),也以酵母染色体为模板扩增获得。作为模板的酵母染色体用拟转化的菌株培养后,用任何市面销售的试剂盒提取即可。本实施例中使用的待转化的酿酒酵母mh001是在重组酵母菌株bsgx001(cen.pk 113
‑
5d derivative;xk,gre3::ppp,cox4δ,ae,pjx7)(wei et al.,2018)基础上,去除其中含有木糖异构酶基因的质粒pjx7而获得的。本实施例中的酵母染色体提取自mh001。
39.本实施例中的荧光报告基因表达盒tef1p
‑
yegfp
‑
pgkt利用引物gfp
‑
tef1
‑
f(seq id no:6)和pgkt
‑
r(seq id no:7),以质粒pjfe1
‑
tef1
‑
gfp(yang et al.,2021)为模板扩增获得。其中,引物gfp
‑
tef1
‑
f包含19bp和35s rdna
‑
1同源的序列,用于片段间的融合;引物pgkt
‑
r包含20bp和表达盒tef1p
‑
kanmx
‑
tef1同源的序列,用于片段间的融合。g418抗性基因表达盒tef1p
‑
kanmx
‑
tef1t利用引物tef1p
‑
f(seq id no:8)和tef1t
‑
r(seq id no:9),以质粒pug6(euroscarf,p30114)为模板扩增获得。木糖异构酶基因表达盒tef1p
‑
ru
‑
xyla
‑
adh1t利用引物adh1t
‑
f(seq id no:10)和引物xi
‑
tef1
‑
r(seq id no:11),以质粒pjx7(wei et al.,2018)为模板扩增获得。其中,引物adh1t
‑
f包含19bp和表达盒tef1p
‑
kanmx
‑
tef1t下游同源的序列,用于片段间的融合;引物xi
‑
tef1
‑
r包含20bp和rdna同源臂2上游同源的序列,用于片段间的融合。
40.将上述35srdna
‑
1、表达盒tef1p
‑
yegfp
‑
pgkt、以及表达盒tef1p
‑
kanmx
‑
tef1t进行融合pcr,使用引物为rdna
‑1‑
f(seq id no:2)和tef1t
‑
r(seq id no:9),获得“融合片段
1”;将上述表达盒tef1p
‑
ru
‑
xyla
‑
adh1t和35srdna
‑
2进行融合pcr,使用引物为adh1t
‑
f(seq id no:10)和rdna
‑2‑
r(seq id no:5),获得“融合片段2”;最后,再将“融合片段1”和“融合片段2”融合,使用引物为rdna
‑1‑
f(seq id no:2)和rdna
‑2‑
r(seq id no:5),最终获得片段rdna
up
‑
gfp
‑
kanmx
‑
ru
‑
xyla
‑
rdna
down
;由于转化需要大量的片段,本实施例中将融合好的片段构建在市售的peasy
‑
blunt克隆载体上,方便以后直接获取转化所需的目的片段,连接方法见peasy
‑
blunt克隆载体说明书。
41.实施例2:常规转化方法获得的转化子荧光强度
42.酿酒酵母培养使用的ypd培养基:20g/l蛋白胨,10g/l酵母粉;固体培养基添加20g/l琼脂粉;灭菌条件:115℃,30分钟。使用时,添加不同浓度的葡萄糖或木糖作为碳源分别制成yepd或yepx培养基。其中,yepd培养基配方:20g/l蛋白胨,10g/l酵母粉,20g/l葡萄糖,若制固体培养基,加入20g/l琼脂粉。yepx培养基配方:20g/l蛋白胨,10g/l酵母粉,10g/l木糖,若制固体培养基,加入20g/l琼脂粉。
43.本实施例中使用的待转化的酿酒酵母mh001是在重组酵母菌株bsgx001(cen.pk113
‑
5d derivative;xk,gre3::ppp,cox4δ,ae,pjx7)(wei et al.,2018)基础上,去除其中含有木糖异构酶基因的质粒pjx7而获得的。
44.酵母转化采用常规醋酸锂(liac)转化法。mh001在20ml yepd培养基中培养12小时后转接至50ml新鲜的yepd培养基,使其初始od
600
值约为0.25。继而在30℃振荡培养至od
600
在0.7
‑
1.0范围区间。5000rpm,离心5分钟收集细胞,无菌水洗涤,再次收集后的细胞重悬于400μl 0.1m liac中混匀。取50μl,13000rpm离心15秒后去上清,然后加入240μl 50%的peg3350,混匀后再加入36μl 1m liac,10μl 10mg/ml的单链鱼精dna(提前100℃煮沸5min后迅速置于冰上保存,1小时内使用),70μl无菌重蒸水溶解的dna片段。
45.本实施例中使用的dna片段为rdna
up
‑
gfp
‑
kanmx
‑
ru
‑
xyla
‑
rdna
down
,利用引物rdna
‑1‑
f(seq id no:2)和rdna
‑2‑
r(seq id no:5),以质粒peasy
‑
blunt
‑
gfp
‑
kanmx
‑
ruxi(seq id no:1)扩增获得。使用量为2μg。上述混合液30℃保温30分钟,42℃热激25分钟,之后8000rpm,离心15秒,去上清。再加入500μl yepd液体培养基,30℃培养2
‑
3小时。之后,该含有转化子的培养液适量涂布在添加20000mg/l g418的yepd的固体培养基平板中,30℃培养2
‑
3天,待转化子长出。
46.随机挑取转化子至含有yepx液体培养基的96孔板中,培养1天转接含有新鲜yepx液体培养基的96孔板,使初始od
600
约等于0.1。用multi
‑
detection microplate reader(synergy ht,biotek,usa)同时读取荧光值和od
600
(即细胞生物量)。相对荧光强度定义为荧光值除以od
600
值。结果显示(图3),96个转化子的平均相对荧光强度为33217rfu,最高相对荧光强度为53370rfu。
47.实施例3:羟基脲预处理降低rdna拷贝
48.菌株mh001在40ml yepd培养基中活化培养12小时后,转接10%菌液到新鲜的含有150mm羟基脲的yepd培养基中培养两天。此后,每两天转接一次,每次都是转接10%菌液到新鲜的含有150mm羟基脲的yepd培养基中。
49.每次转接之后将剩余的细胞提取其染色体,并用荧光定量pcr法测定其rdna拷贝数。结果显示,初始mh001细胞群体,rdna拷贝数为144.7
±
28.6个拷贝/细胞。经过2、4、6和8天在含有羟基脲的培养基中的培养处理,拷贝分别降至132.9
±
18.5、101.4
±
16.7、83.5
±
2.4和81.0
±
8.3个拷贝/细胞(图4)。其中第八天的培养物中,分离出的细胞,命名为mh001
‑
8d。
50.实施例4:羟基脲预处理后的菌株转化dna获得的转化子的荧光强度
51.用实施例2中同样的dna片段,采取实施例2中完全相同的方法转化菌株mh001
‑
8d。转化子一部分涂布在添加20000mg/l g418的yepd的固体培养基平板中(图1中的方案一),另一部分涂布在添加20000mg/l g418和60mm羟基脲的yepd的固体培养基平板中(图1中的方案二),30℃培养2
‑
3天,待转化子长出。
52.采取实施例2中的方案,测定转化子的荧光强度。
53.在方案一和方案二的平板上各随机挑取转化子至含有yepx液体培养基的96孔板中,培养1天转接含有新鲜yepx液体培养基的96孔板,使初始od
600
约等于0.1。用multi
‑
detection microplate reader(synergy ht,biotek,usa)同时读取荧光值和od
600
(即细胞生物量)。相对荧光强度定义为荧光值除以od
600
值。
54.结果显示,方案一中随机挑选的96个转化子的平均相对荧光强度为32834rfu,最高相对荧光强度为54370rfu(图5);方案二中随机挑选的96个转化子的平均相对荧光强度为35068rfu,最高相对荧光强度为71156rfu(图6),虽然平均相对荧光强度提高不显著,但最高荧光强度均有所提高。
55.实施例5:各种方法获得的重组菌株中ru
‑
xyla拷贝数
56.选取实施例2中获得的转化子中荧光强度最高的5株,实施例4中方案一获得的转化子中荧光强度最高的5株,以及实施例4中方案二获得的转化子中荧光强度最高的5株,用荧光定量pcr方法检测ru
‑
xyla基因拷贝数。
57.结果显示(图7),实施例2中获得的5株转化子ru
‑
xyla基因平均拷贝数为7.7,最高拷贝数为10.1;实施例4中方案一中获得的5株转化子ru
‑
xyla基因平均拷贝数为9.0,最高拷贝数为10.3;实施例4中方案二中获得的5株转化子ru
‑
xyla基因平均拷贝数为13.1,最高拷贝数为18.0。这一结果表明,羟基脲预处理菌株后再转化目的基因能够提高外源基因整合的拷贝数。其中,实施例4中方案二为更优方案。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1