一种平面手性噁唑啉-醇二茂铁化合物及其制备、应用
一种平面手性噁唑啉
‑
醇二茂铁化合物及其制备、应用
技术领域
1.本发明属于化合物合成技术领域,具体涉及一种具有硅醚取代基的平面手性噁唑啉
‑
醇二茂铁化合物及其制备、应用。
背景技术:
2.手性二茂铁
‑
醇化合物被认为是不对称醛、酮化合物亲核加成反应过程中重要的手性催化剂类型之一。作为手性二茂铁
‑
醇类催化剂的重要分支,手性噁唑啉
‑
醇二茂铁化合物在多种亲核加成反应中被成功应用并催化得到高立体选择性的手性醇产物。
3.在手性噁唑啉二茂铁化合物进行不对称反应应用时,对噁唑啉手性侧链取代基的筛选是重要的反应条件优化过程。传统手性噁唑啉二茂铁化合物的手性取代基来源于合成噁唑啉环的手性氨基醇类化合物。这意味着噁唑啉环手性侧链局限于氨基酸及其相应氨基醇的化合物结构。目前常见的噁唑啉手性侧链基团,为异丙基、叔丁基、苯基及苄基基团。
4.为进一步扩展噁唑啉环手性侧链结构,并使噁唑啉二茂铁类化合物更适用于复杂反应底物的不对称转化过程,对噁唑啉手性侧链衍生的相关工作不断被报道。2015年,周智明课题组报道了一种新型噁唑啉手性侧链结构,将硅醚基团引入噁唑啉环手性侧链。随后该课题组将合成的噁唑啉
‑
膦化合物作为手性配体应用于多种过渡金属催化的不对称反应中。上述提及的化合物主要用于钯、铜、银催化的不对称催化反应过程,但是并不能满足锌参与的不对称醛、酮化合物加成反应。本发明希望合成一种用于锌参与的不对称醛、酮化合物加成反应的化合物,期望催化得到高立体选择性的手性醇化合物。
技术实现要素:
5.有鉴于此,本发明的目的在于提供一种具有硅醚取代基的平面手性噁唑啉
‑
醇二茂铁化合物及其制备、应用。
6.本发明所采用的技术方案为:
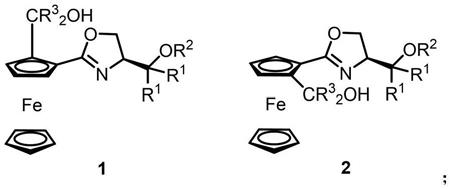
7.一种平面手性噁唑啉
‑
醇二茂铁化合物,具有如式1或式2的结构:
[0008][0009]
其中,r1、r3分别独立选自氢、烷基、苯基、取代苯基或二茂铁基中的一种;
[0010]
r2选自三甲基硅基、三乙基硅基、叔丁基二甲基硅基、三苯基硅基或由其他烷基取代硅基中的一种;
[0011]
噁唑啉手性侧链中心手性为(s)构型;
[0012]
式1化合物中二茂铁化合物中平面手性为(sp)构型,式2化合物中二茂铁化合物中平面手性为(rp)构型。
[0013]
制备平面手性噁唑啉
‑
醇二茂铁化合物的方法,包括以下步骤:
[0014]
1)以(s)
‑4‑
甲酯噁唑啉
‑2‑
基二茂铁(化合物3)为原料,以醚类有机溶剂或卤代烃类溶剂为溶剂,在还原剂作用下,反应在
‑
10~30℃条件下,将噁唑啉环侧链酯基还原为醇化合物,得到噁唑啉环侧链酯基还原醇(化合物4);
[0015]
2)以步骤1)制得的噁唑啉环侧链酯基还原醇(化合物4)为原料,以醚类有机溶剂为溶剂,与烷基氯硅烷反应,反应在
‑
10~30℃条件下,得到噁唑啉二茂铁硅醚化合物(化合物5);
[0016]
3)以步骤2)制得的噁唑啉二茂铁硅醚化合物(化合物5)为原料,在高纯氮气保护下,以无水无氧乙醚为溶剂,在配体的协同作用下,于
‑
78~
‑
80℃条件下,向反应体系中加入正丁基锂的正己烷溶液;在
‑
78~
‑
80℃条件反应2小时后,加入酮类化合物,将反应逐渐升温至
‑
10~30℃条件,得到噁唑啉
‑
醇二茂铁化合物;
[0017]
当配体为n,n,n,n
‑
四甲基乙二胺(tmeda)时,得到平面手性为(sp)构型的噁唑啉
‑
醇二茂铁化合物;
[0018]
当配体为二乙二醇二醚(dgme)时,得到平面手性为(rp)构型的噁唑啉
‑
醇二茂铁化合物。
[0019]
所述醚类有机溶剂为无水乙醚、四氢呋喃或1,4
‑
二氧六环;所述卤代烃类溶剂为一氯甲烷、二氯甲烷或三氯甲烷。
[0020]
所述还原剂为四氢铝锂、硼氢化钠或格式试剂,所述格式试剂为烷基、苯基或取代苯基的格式试剂。
[0021]
当还原剂为四氢铝锂、硼氢化钠时,r1为氢;当还原剂为格式试剂时,r1为格式试剂中烷基、苯基或取代苯基等取代基。
[0022]
所述烷基氯硅烷选自三甲基氯硅烷、三乙基氯硅烷、叔丁基二甲基氯硅烷、三苯基氯硅烷或其他烷基取代氯硅烷。
[0023]
所述酮类化合物为二苯甲酮、多聚甲醛、丙酮或双二茂铁甲酮。
[0024]
所述二乙二醇二醚的结构为
[0025]
所述二乙二醇二醚结构中的r为c1
‑
c6烷基、苯基或取代苯基的一种。
[0026]
噁唑啉
‑
醇二茂铁化合物在不对称醛、酮化合物加成反应中作为催化剂的应用。
[0027]
本发明平面手性为(sp)构型的噁唑啉
‑
醇二茂铁化合物的制备路线如下:
[0028]
[0029]
当配体为二乙二醇二醚制备平面手性为(rp)构型的噁唑啉
‑
醇二茂铁化合物时的最后一步制备路线为:
[0030][0031]
与现有技术相比,本发明的有益技术效果是:
[0032]
1、本发明相对传统含有烷基或芳基手性侧链的噁唑啉
‑
醇二茂铁化合物,其手性侧链更易于广泛衍生,获得的噁唑啉
‑
醇二茂铁硅醚化合物在亲核加成反应,尤其是在锌参与的不对称醛、酮化合物加成反应中具有突出的催化效果。
[0033]
2、本发明的噁唑啉
‑
醇二茂铁化合物在制备时,采用了基于酮类化合物为亲电试剂的二茂铁化合物平面手性引入的新方法,这样的平面手性引入方法更加简便。
具体实施方式
[0034]
下面结合实施例来说明本发明的具体实施方式,但以下实施例只是用来详细说明本发明,并不以任何方式限制本发明的范围。对本领域的技术人员来说,在不脱离本发明构思的前提下,亦可作出若干合成路线的改变和技术改进。以上改变和技术改进均属于本发明的保护范围。
[0035]
实施例1:
[0036]
(s)
‑4‑
(羟基二乙基)甲基
‑
噁唑啉
‑2‑
基二茂铁(4a):
[0037]
在高纯氮气保护下,在250ml反应瓶中加入1.21g(s)
‑4‑
甲酯噁唑啉
‑2‑
基二茂铁(3),随后加入80ml无水乙醚;待全部固体溶解后,将反应体系降温至0℃;向反应瓶中加入15.5ml乙基溴化镁的四氢呋喃溶液(浓度1.0m),滴加过程持续45分钟,反应保持在0℃条件下进行。利用薄层色谱法检测原料全部转化后,在0℃条件下加入200ml去离子水淬灭反应,并用乙醚萃取水相中的有机物直至水层无色;合并油相,无水硫酸镁干燥;将油相浓缩旋干溶剂,柱层析分离(洗脱剂石油醚:乙酸乙酯=5:1)得到产物(s)
‑4‑
(羟基二乙基)甲基
‑
噁唑啉
‑2‑
基二茂铁(4a)。
[0038]
产物结构式:
[0039]
产物性状为橙色固体,产率86%,熔点:80
‑
81℃,旋光度[α]
d20
=+100.7(浓度1.0g/100ml,溶剂二氯甲烷);核磁氢谱(600mhz,溶剂氘代氯仿)δ4.77(br s,1h,cp
‑
h),4.74(br s,1h,cp
‑
h),4.34(br s,2h,cp
‑
h
×
2),4.33
‑
4.28(m,2h,ochh+ochh),4.22
‑
4.21(m,1h,chn),4.19(s,5h,cp
‑
h
×
5),1.79
‑
1.75(m,2h,ch2ch3),1.56
‑
1.51(m,2h,ch2ch3),1.38
‑
1.35(m,1h,cet2oh),0.96(t,j=7.5hz,3h,ch2ch3),0.92(t,j=7.5hz,3h,ch2ch3);产物核磁碳谱(151mhz,溶剂氘代氯仿)δ167.6(c=n),75.0,72.5,70.3,69.6(cp
‑
c
×
5),69.1(d,j=7hz),67.9,28.6,26.4,7.8,7.6;高分辨质谱:(esi
‑
tof)计算值c
18
h
23
feno2[m+
h]
+
:342.1151;实际测量值342.1133。
[0040]
(s)
‑4‑
(三甲基硅氧基二乙基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5a):
[0041]
在250ml单口瓶中加入1.02g(s)
‑4‑
(羟基二乙基)甲基
‑
噁唑啉
‑2‑
基二茂铁(4a),1.02g咪唑,随后加入50ml二氯甲烷;待全部固体溶解后,将反应体系降温至0℃;向单口瓶中加入650mg三甲基氯硅烷,反应液逐渐变浑浊;室温反应过夜后,加入去离子水淬灭反应,并用无水乙醚萃取反应液,直至水层无色;合并油相,无水硫酸镁干燥;将油相浓缩旋干溶剂,柱层析分离(洗脱剂石油醚:乙酸乙酯=15:1)得到产物(s)
‑4‑
(三甲基硅氧基二乙基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5a)。
[0042]
产物结构式:
[0043]
产物性状为橙红色固体,产率76%,熔点:91
‑
92℃,旋光度[α]
d20
=
‑
18.3(浓度1.0g/100ml,溶剂二氯甲烷);核磁氢谱(600mhz,溶剂氘代氯仿)δ4.73(br s,1h,cp
‑
h),4.69(br s,1h,cp
‑
h),4.33
‑
4.30(m,3h,cp
‑
h
×
2+ochh),4.25
‑
4.23(m,1h,ochh),4.19(s,5h,cp
‑
h
×
5),4.19
‑
4.16(m,1h,chn),1.90
‑
1.84(m,1h,chhch3),1.67
‑
1.61(m,1h,chhch3),1.51
‑
1.46(m,2h,chhch3×
2),0.94
‑
0.90(dt,6h,ch2ch3×
2),0.08(s,9h,si(ch3)3);产物核磁碳谱(151mhz,溶剂氘代氯仿)δ167.7(c=n),79.9,73.3,70.0,69.8,69.5(cp
‑
c
×
5),68.9(d,j=7hz),68.1,29.6,27.9,8.7,8.0,2.8(si(ch3)3);高分辨质谱:(esi
‑
tof)计算值c
21
h
31
feno2si[m+h]
+
:414.1545;实际测量值414.1511。
[0044]
实施例2
[0045]
(s)
‑4‑
(羟基二苯基)甲基
‑
噁唑啉
‑2‑
基二茂铁(4b):
[0046]
在高纯氮气保护下,在500ml反应瓶中加入1.44g(s)
‑4‑
甲酯噁唑啉
‑2‑
基二茂铁(3),随后加入110ml无水乙醚;待全部固体溶解后,将反应体系降温至0℃;向反应瓶中加入18.8ml苯基溴化镁的四氢呋喃溶液(浓度1.0m),滴加过程持续60分钟,反应保持在0℃条件下进行。利用薄层色谱法检测原料全部转化后,在0℃条件下加入250ml去离子水,并用乙醚萃取水相中的有机物直至水层无色;合并油相,无水硫酸镁干燥;将油相浓缩旋干溶剂,柱层析分离(洗脱剂石油醚:乙酸乙酯=12:1)得到产物(s)
‑4‑
(羟基二苯基)甲基
‑
噁唑啉
‑2‑
基二茂铁(4b)。
[0047]
产物结构式:
[0048]
产物性状为橙色固体,产率78%,熔点:186
‑
188℃,旋光度[α]
d20
=
‑
28.4(浓度1.0g/100ml,溶剂二氯甲烷);核磁氢谱(600mhz,溶剂氘代氯仿)δ7.66
‑
7.64(m,2h,ph
‑
h),7.47
‑
7.45(m,2h,ph
‑
h),7.37
‑
7.30(m,5h,ph
‑
h),7.24
‑
7.20(m,1h,ph
‑
h),5.34(t,j=9.3hz,1h,chn),4.74(br s,1h,cp
‑
h),4.71(br s,1h,cp
‑
h),4.32(br s,2h,cp
‑
h
×
2),4.19(s,5h,cp
‑
h
×
5),4.16
‑
4.14(m,2h,ochh+ochh);产物核磁碳谱(151mhz,溶剂氘代氯仿)δ169.6(c=n),146.2,144.3,128.3,128.2,127.0,126.9,125.8,78.0,73.0,71.1,70.4
(d,j=14hz),70.0,69.7(cp
‑
c
×
5),69.4,69.2,69.0;高分辨质谱:(esi
‑
tof)计算值c
26
h
23
feno2[m+h]
+
:438.1511;实际测量值438.1120。
[0049]
(s)
‑4‑
(三甲基硅氧基二苯基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5b):
[0050]
在250ml单口瓶中加入1.16g(s)
‑4‑
(羟基二苯基)甲基
‑
噁唑啉
‑2‑
基二茂铁(4b),903mg咪唑,随后加入45ml二氯甲烷;待全部固体溶解后,将反应体系降温至0℃;向单口瓶中加入577mg三甲基氯硅烷,反应液逐渐变浑浊;室温反应过夜后,加入去离子水淬灭反应,并用无水乙醚萃取反应液,直至水层无色;合并油相,无水硫酸镁干燥;将油相浓缩旋干溶剂,柱层析分离(洗脱剂石油醚:乙酸乙酯=15:1)得到产物(s)
‑4‑
(三甲基硅氧基二苯基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5b)。
[0051]
产物结构式:产物性状为橙色固体,产率62%,熔点大于250℃,旋光度[α]
d20
=
‑
15.6(浓度1.0g/100ml,溶剂二氯甲烷);核磁氢谱(600mhz,溶剂氘代氯仿)δ7.53
‑
7.50(m,4h,ph
‑
h),7.31
‑
7.29(m,5h,ph
‑
h),7.21
‑
7.18(m,1h,ph
‑
h),5.24(t,j=5.7hz,1h,chn),4.66(br s,1h,cp
‑
h),4.58(br s,1h,cp
‑
h),4.26
‑
4.25(m,2h,cp
‑
h
×
2),4.22
‑
4.18(m,2h,ochh+ochh),4.03(s,5h,cp
‑
h
×
5),
‑
0.15(si(ch3)3);产物核磁碳谱(151mhz,溶剂氘代氯仿)δ167.2(c=n),146.5,144.4,129.0,128.0,127.8,127.1,127.0,126.8,82.0,73.6,71.2,69.9(d,j=16hz),69.5(cp
‑
c
×
5),69.0,68.6,1.9(si(ch3)3);高分辨质谱:(esi
‑
tof)计算值c29h31feno2si[m+h]+:510.1547;实际测量值510.1566。
[0052]
实施例3
[0053]
(s)
‑4‑
(叔丁基二甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5c):
[0054]
1)将(r)
‑4‑
羟甲基
‑2‑
二茂铁基噁唑啉(1.0g,3.5mmol)和咪唑(1.07g,15.8mmol)溶于35毫升无水四氢呋喃中,随后加入二甲基叔丁基氯硅烷(1.05g,7mmol),反应液立刻变浑浊。室温反应过夜后,加入大量乙醚和饱和食盐水,无水乙醚萃取反应液,直至水层无色,合并油相,无水硫酸钠干燥。蒸出溶剂,粗产品柱层析分离(硅胶100
‑
200目,洗脱剂:石油醚:乙酸乙酯=15:1)。产物(s)
‑4‑
(叔丁基二甲基硅氧基二甲基硅氧基)甲基
‑
噁唑啉
‑2‑
基二茂铁(4c),深红色液体,产率93%。[α]
d20
=+24.1(c 0.28,ch2cl2)。1h nmr(400mhz,cdcl3):δ0.07(s,6h,si(ch3)2),0.90(s,9h,c(ch3)3),3.56(dd,j=7.2,6.4hz,1h,chhotbs),3.88(dd,j=11.8,3.6hz,1h,chhotbs),4.12(s,5h,cp
‑
h),4.22
‑
4.33(m,5h,och2+chn+cp
‑
h
×
2),4.73(br s,2h,cp
‑
h)。
13
c nmr(100mhz,d6‑
dmso)δ
‑
5.03,18.05,25.08,64.67,67.66,68.65(d,j=17hz),69.44(cp
×
5),70.01,70.33,165.36。ms(esi)m/z(m+h
+
)400.5。
[0055]
实施例4
[0056]
(s)
‑4‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5d):
[0057]
将(s)
‑4‑
(羟基二甲基)甲基
‑2‑
二茂铁基噁唑啉(300mg,0.96mmol)和咪唑(326mmol,4.8mmol)溶于20毫升无水四氢呋喃中,随后加入三甲基氯硅烷(287mg,1.92mmol),反应液立刻变浑浊。室温反应过夜后,加入大量乙醚和饱和食盐水,无水乙醚萃取反应液,直至水层无色,合并油相,无水硫酸钠干燥。蒸出溶剂,粗产品柱层析分离(硅胶
100
‑
200目,洗脱剂:石油醚:乙酸乙酯=20:1)。产物(s)
‑4‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基二茂铁,橙色固体,产率93%。m.p.:59
‑
60℃。ir(cm
‑1):3088,3071,2977,2932,2898,1649,1250,1164,1114,1017。1h
‑
nmr(400mhz,cdcl3):δ0.11(9h,s,si(ch3)3),1.20(3h,s,c(ch3)(ch3)otms),1.38(3h,s,c(ch3)(ch3)oh),4.04(2h,m,cp
‑
h),4.22(5h,s,cp
‑
h),4.25(1h,m,chho),4.32(1h,br s,chn),4.39(1h,m,chho),4.71(1h,br s,cp
‑
h),4.75(1h,br s,cp
‑
h)。ms(esi)m/z(m+h
+
)386.3。
[0058]
实施例5
[0059]
在高纯氮气保护下,在100ml干燥反应瓶中加入970mg(s)
‑4‑
(叔丁基二甲基硅氧基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5c,实施例3制得),高纯氮气置换三次;加入15ml无水无氧处理后乙醚,将原料完全溶解;随后向体系中加入338mg新蒸n,n,n,n
‑
四甲基乙二胺。利用冰丙酮浴将反应体系降温至
‑
78℃,向体系滴加1.8ml正丁基锂(1.6m),15分钟加毕;滴加完毕后,将体系缓慢恢复至0℃,反应2小时,反应体系由橙色逐渐变为红色;再次将体系降温至
‑
78℃后,向体系加入530mg重结晶二苯甲酮,体系逐渐变蓝;将体系缓慢恢复至室温,反应过夜,溶液中有沉淀析出。反应完毕,向体系中加入冰的饱和氯化铵水溶液淬灭反应,体系由蓝绿色变为橙黄色;乙酸乙酯萃取反应液,直至水层无色;合并油相,无水硫酸镁干燥;将油相浓缩旋干溶剂,柱层析分离(洗脱剂石油醚:乙酸乙酯=100:1)得到产物(s,sp)
‑1‑
[4
‑
(叔丁基二甲基硅氧基)甲基
‑
噁唑啉
‑2‑
基]
‑2‑
羟基二苯基甲基二茂铁(1a)。
[0060]
产物结构式:产物性状为黄色固体,产率80%,熔点:161
‑
162℃,旋光度[α]
d20
=+241.9(浓度0.17g/100ml,溶剂二氯甲烷);核磁氢谱(400mhz,溶剂氘代氯仿)δ9.05(s,1h,cph2oh),7.54
‑
7.52(m,2h,ph
‑
h),7.36
‑
7.32(m,2h,ph
‑
h),7.28(m,1h,ph
‑
h),7.19
‑
7.13(m,5h,ph
‑
h),4.78(s,1h,cp
‑
h),4.29(s,5h,cp
‑
h
×
5),4.28
‑
4.24(m,2h,chn+cp
‑
h),4.19(t,j=8.8hz,1h,chhotbs),4.00
‑
3.95(m,1h,ochh),3.86(dd,j=10.1,4.0hz,1h,chhotbs),3.73(s,1h,cp
‑
h),3.62(dd,j=10.0,7.1hz,1h,ochh),0.95(s,9h,c(ch3)3),0.14(s,6h,si(ch3)2);产物核磁碳谱(101mhz,溶剂氘代氯仿)δ169.1(c=n),149.4(ph
‑
c),146.5(ph
‑
c),127.9(ph
‑
c),127.5(ph
‑
c),127.1(ph
‑
c),127.0(ph
‑
c),126.5(ph
‑
c),126.3(ph
‑
c),100.5(cph2oh),77.2,75.1,71.0(cp
‑
c
×
5),70.5,69.8,68.1,67.2,65.7,64.8,26.0,18.4,
‑
5.27;高分辨质谱:(esi
‑
tof)计算值c
33
h
40
feno3si[m+h]
+
:582.2122;实际测量值582.2127。
[0061]
实施例6
[0062]
在高纯氮气保护下,在50ml干燥反应瓶中加入800mg(s)
‑4‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5d,实施例4制得),高纯氮气置换三次;加入15ml无水无氧处理后乙醚,将原料完全溶解;随后向体系中加入314mg新蒸n,n,n,n
‑
四甲基乙二胺。利用冰丙酮浴将反应体系降温至
‑
78℃,向体系滴加1.7ml正丁基锂(1.6m),15分钟加毕;滴加完毕后,将体系缓慢恢复至0℃,反应1.5小时,反应体系由橙色逐渐变为橙红色;再次将体系降温至
‑
78℃后,向体系加入492mg重结晶二苯甲酮,体系变为深蓝色;将体系缓慢恢复至室
温,反应过夜,溶液中有沉淀析出。反应完毕,向体系中加入冰的饱和氯化铵水溶液,体系由蓝绿色变为橙黄色;无水乙醚萃取反应液,直至水层无色;合并油相,无水硫酸镁干燥;将油相浓缩旋干溶剂,柱层析分离(洗脱剂石油醚:乙酸乙酯=40:1)得到产物(s,sp)
‑1‑
[4
‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基]
‑2‑
羟基二苯基甲基二茂铁(1b)。
[0063]
产物结构式:
[0064]
产物性状为橙黄色固体,产率81%,熔点:131
‑
132℃,旋光度[α]
d20
=+247.5(浓度1.0g/100ml,溶剂二氯甲烷);核磁氢谱(600mhz,溶剂氘代氯仿)δ9.08(br s,cph2oh),7.52
‑
7.31(m,10h,ph
‑
h),4.73(pst,1h,cp
‑
h),4.28(s,5h,cp
‑
h
×
5),4.27
‑
4.24(m,2h,chn+cp
‑
h),4.14
‑
4.09(m,1h,ochh),3.69
‑
3.65(m,2h,ochh+cp
‑
h),1.33(s,3h,ch3),1.29(s,3h,ch3),0.15(s,9h,si(ch3)3);产物核磁碳谱(151mhz,溶剂氘代氯仿)δ168.5(c=n),149.2,146.4,128.1,127.9,127.4,127.1(d,j=4hz),126.7,126.6,126.2,126.0,100.6,77.3,75.7,75.0,74.2,70.7(cp
‑
c
×
5),70.4,68.6,68.0,66.0,28.8(ch3),25.5(ch3),2.6(si(ch3)3);高分辨质谱:(esi
‑
tof)计算值c
32
h
37
feno3si[m+h]
+
:568.1992;实际测量值568.2000。
[0065]
实施例7
[0066]
在高纯氮气保护下,在50ml干燥反应瓶中加入878mg(s)
‑4‑
(三甲基硅氧基二乙基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5a,实施例1制得),高纯氮气置换三次;加入15ml无水无氧处理后乙醚,将原料完全溶解;随后向体系中加入309mg新蒸n,n,n,n
‑
四甲基乙二胺。利用冰丙酮浴将反应体系降温至
‑
78℃,向体系滴加1.7ml正丁基锂(浓度1.6m),15分钟加毕;滴加完毕后,将体系缓慢恢复至0℃,反应4小时,反应体系由橙色逐渐变为橙红色;再次将体系降温至
‑
78℃后,向体系加入485mg重结晶二苯甲酮,体系变为深蓝色;将体系缓慢恢复至室温,反应过夜,溶液中有沉淀析出。反应完毕,向体系中加入冰的饱和氯化铵水溶液,体系由蓝绿色变为橙黄色;无水乙醚萃取反应液,直至水层无色;合并油相,无水硫酸镁干燥;将油相浓缩旋干溶剂,柱层析分离(洗脱剂石油醚:乙酸乙酯=40:1)得到产物(s,sp)
‑1‑
[4
‑
(三甲基硅氧基二乙基)甲基
‑
噁唑啉
‑2‑
基]
‑2‑
羟基二苯基甲基二茂铁(1c)。
[0067]
产物结构式:产物性状为橙红色晶体,产率72%,熔点:130
‑
131℃,旋光度[α]
d20
=+144.7(浓度0.17g/100ml,溶剂二氯甲烷);核磁氢谱(600mhz,溶剂氘代氯仿)δ8.99(s,1h,cph2oh),7.50(dd,j=15.2,4.6hz,2h,ph
‑
h
×
2),7.44
–
7.39(m,h,ph
‑
h),7.31(dt,j=7.6,3.5hz,3h,ph
‑
h
×
3),7.14(q,j=4.9,3.4hz,4h,ph
‑
h
×
4),4.73(s,1h,cp
‑
h),4.32(s,5h,cp
‑
h
×
5),4.27(s,1h,ochh),4.24(t,j=2.6hz,1h,cp
‑
h),4.11(t,j=9.0hz,1h,chn),3.73(t,j=10.0hz,1h,ochh),3.67(s,1h,cp
‑
h),1.93(dq,j=14.6,7.4hz,1h,ch2ch3),1.68(dq,j=15.5,8.3,7.8hz,1h,ch2ch3),1.58(dt,j=14.8,
7.4hz,1h,ch2ch3),1.46(dt,j=14.9,7.5hz,1h,ch2ch3),0.90
–
0.88(m,3h,ch2ch3),0.84(t,j=7.5hz,3h,ch2ch3),0.18(s,9h,si(ch3)3).产物核磁碳谱(151mhz,溶剂氘代氯仿)δ167.92(c=n),128.08(ph
‑
c
×
2),127.79(ph
‑
c
×
2),127.40(ph
‑
c),127.09(ph
‑
c
×
2),126.70(ph
‑
c),126.54(ph
‑
c),126.16(ph
‑
c),125.99(ph
‑
c
×
2),100.90,79.07,78.22,74.97,72.30,70.69(cp
‑
c
×
5),70.23,68.26,67.80,31.03(ch2ch3),30.86(ch2ch3),8.61(ch2ch3),8.40(ch2ch3),2.81(si(ch3)3).高分辨质谱:(esi
‑
tof)计算值c
34
h
42
feno3si[m+h]
+
:596.2278;测量值596.2285。
[0068]
实施例8
[0069]
在高纯氮气保护下,在100ml干燥反应瓶中加入300mg(s)
‑4‑
(三甲基硅氧基二苯基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5b,实施例2制得),高纯氮气置换三次;加入40ml无水无氧处理后乙醚,将原料完全溶解;随后向体系中加入429mg新蒸n,n,n,n
‑
四甲基乙二胺。利用冰丙酮浴将反应体系降温至
‑
78℃,向体系滴加2.8ml正丁基锂(1.6m),15分钟加毕;滴加完毕后,将体系缓慢恢复至0℃,反应6小时,反应体系由橙色逐渐变为橙红色;再次将体系降温至
‑
78℃后,向体系加入650mg重结晶二苯甲酮;将体系缓慢恢复至室温,反应过夜,溶液中有沉淀析出。反应完毕,向体系中加入冰的饱和氯化铵水溶液,体系由蓝绿色变为橙黄色;无水乙醚萃取反应液,直至水层无色;合并油相,无水硫酸镁干燥;将油相浓缩旋干溶剂,柱层析分离(洗脱剂石油醚:乙酸乙酯=150:1)得到产物(s,sp)
‑1‑
[4
‑
(三甲基硅氧基二苯基)甲基
‑
噁唑啉
‑2‑
基]
‑2‑
(羟基二苯基)甲基二茂铁(1d)。
[0070]
产物结构式:
[0071]
产物性状为橙色固体,产率33%,熔点大于230℃(分解),旋光度[α]
d20
=+79.4(浓度0.15/100ml,溶剂二氯甲烷);核磁氢谱(600mhz,溶剂氘代氯仿)δ8.50(s,1h,cph2oh),7.42
–
7.39(m,6h,ph
‑
h
×
6),7.30(d,j=7.9hz,5h,ph
‑
h
×
5),7.27(d,j=10.0hz,5h,ph
‑
h
×
5),7.22(t,j=7.3hz,4h,ph
‑
h
×
4),4.82(t,j=10.5hz,1h,chn),4.59(s,1h,cp
‑
h),4.30(t,j=9.3hz,1h,ochh),4.23
–
4.14(m,2h,ochh+cp
‑
h),3.96(s,5h,cp
‑
h
×
5),3.61(s,1h,cp
‑
h),0.07(s,9h,si(ch3)3).产物核磁碳谱(151mhz,溶剂氘代氯仿)δ147.16(c=n),143.75,129.00
–
128.84(m)(ph
‑
c
×
6),128.09(ph
‑
c
×
4),127.85(ph
‑
c
×
2),127.73(ph
‑
c
×
2),127.08(ph
‑
c
×
4),126.71(ph
‑
c
×
2),125.99(ph
‑
c
×
4),84.71,82.12,81.64,78.24,75.30,72.53,70.52(cp
‑
c
×
5),70.44,1.01(si(ch3)3).高分辨质谱:(esi
‑
tof)计算值c
42
h
42
feno3si[m+h]
+
:692.2279;测量值692.2302。
[0072]
实施例9
[0073]
在高纯氮气保护下,在50ml干燥反应瓶中加入800mg(s)
‑4‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5d,实施例4制得),高纯氮气置换三次;加入15ml无水无氧处理后乙醚,将原料完全溶解;随后向体系中加入363mg新蒸n,n,n,n
‑
四甲基乙二胺。利用冰丙酮浴将反应体系降温至
‑
78℃,向体系滴加1.95ml正丁基锂(1.6m),15分钟加毕;滴加完毕后,将体系缓慢恢复至0℃,反应3小时,反应体系由橙色逐渐变为橙红色;再次将体系降
温至
‑
78℃后,向体系加入609mg新蒸馏n,n
‑
二甲基甲酰胺;将体系缓慢恢复至室温,反应6小时后,在0℃条件下分5次共加入473mg硼氢化钠,溶液中有沉淀析出。反应完毕,向体系中加入冰水淬灭反应,并用乙酸乙酯萃取反应液,直至水层无色;合并油相,无水硫酸镁干燥;将油相浓缩旋干溶剂,柱层析分离(洗脱剂石油醚:乙酸乙酯=50:1)得到产物(s,sp)
‑1‑
[4
‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基]
‑2‑
羟甲基二茂铁(1e)。
[0074]
产物结构式:
[0075][0076]
产物性状为橙色固体,产率86%,熔点:116
‑
117℃,旋光度[α]
d20
=+156.5(浓度1.0g/100ml,溶剂二氯甲烷);核磁氢谱(600mhz,溶剂氘代氯仿)δ4.70
–
4.57(m,2h,cp
‑
h
×
2),4.46(t,j=7.9hz,1h,chn),4.34(s,2h,ch2oh),4.30(t,j=9.1hz,1h,ochh),4.24(s,1h,cp
‑
h),4.21(s,5h,cp
‑
h
×
5),4.07
–
4.01(m,1h,ochh),1.36(s,3h,ch3),1.32(s,3h,ch3),0.17(s,9h,si(ch3)3).产物核磁碳谱(151mhz,溶剂氘代氯仿)δ168.51(c=n),75.82,74.43,72.00,70.28,69.92(cp
‑
c
×
5),69.12,68.08,28.64(ch3),25.01(ch3),2.54(si(ch3)3).高分辨质谱:(esi
‑
tof)计算值c
20
h
30
feno3si[m+h]
+
:416.1339;测量值416.1329。
[0077]
实施例10
[0078]
在高纯氮气保护下,在50ml干燥反应瓶中加入450mg(s)
‑4‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5d,实施例4制得),高纯氮气置换三次;加入18ml无水无氧处理后乙醚,将原料完全溶解;随后向体系中加入170mg新蒸n,n,n,n
‑
四甲基乙二胺。利用冰丙酮浴将反应体系降温至
‑
78℃,向体系滴加0.9ml正丁基锂(1.6m),10分钟加毕;滴加完毕后,将体系缓慢恢复至0℃,反应3小时,反应体系由橙色逐渐变为橙红色;再次将体系降温至
‑
78℃后,向体系加入86mg新蒸馏无水丙酮;将体系缓慢恢复至室温,反应8小时;反应完毕,向体系中加入冰的饱和氯化铵水溶液;并用乙酸乙酯萃取反应液,直至水层无色;合并油相,无水硫酸镁干燥;将油相浓缩旋干溶剂,柱层析分离(洗脱剂石油醚:乙酸乙酯=80:1)得到产物(s,sp)
‑1‑
[4
‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基]
‑2‑
(羟基二甲基)甲基二茂铁(1f)。
[0079]
产物结构式:
[0080]
产物性状为橙色固体,产率90%,熔点:85
‑
87℃,旋光度[α]
d20
=+180.6(浓度1.0g/100ml,溶剂二氯甲烷);核磁氢谱(600mhz,溶剂氘代氯仿)δ7.90(s,1h,cph2oh),
4.66
–
4.62(m,1h,cp
‑
h),4.40(t,j=8.3hz,1h,chn),4.28(s,7h,cp
‑
h
×
5+cp
‑
h+ochh),4.23(t,j=2.5hz,1h,ochh),4.06(dd,j=9.9,8.2hz,1h,cp
‑
h),1.64(s,3h,ch3),1.40(s,3h,ch3),1.37(s,3h,ch3),1.32(s,3h,ch3),0.15(s,9h,si(ch3)3).产物核磁碳谱(151mhz,溶剂氘代氯仿)δ169.30(c=n),100.58,76.03,74.43,70.41,70.25(cp
‑
c
×
5),68.90(d,j=2.3hz),68.15,65.29,32.44(ch3),29.44(ch3),28.94(ch3),25.10(ch3),2.56(si(ch3)3).高分辨质谱:(esi
‑
tof)计算值c
22
h
34
feno3si[m+h]
+
:444.1652;实际测量值444.1650。
[0081]
实施例11
[0082]
在高纯氮气保护下,在50ml干燥反应瓶中加入564mg(s)
‑4‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5d,实施例4制得),高纯氮气置换三次;加入15ml无水无氧处理后乙醚,将原料完全溶解;随后向体系中加入213mg新蒸n,n,n,n
‑
四甲基乙二胺。利用冰丙酮浴将反应体系降温至
‑
78℃,向体系滴加1.2l正丁基锂(1.6m),15分钟加毕;滴加完毕后,将体系缓慢恢复至0℃,反应4小时,反应体系由橙色逐渐变为橙红色;再次将体系降温至
‑
78℃后,向体系加入728mg双二茂铁甲酮;将体系缓慢恢复至室温,反应过夜;反应完毕,向体系中加入冰的饱和氯化铵水溶液;无水乙醚萃取反应液,直至水层无色;合并油相,无水硫酸镁干燥;将油相浓缩旋干溶剂,柱层析分离(洗脱剂石油醚:乙酸乙酯=50:1)得到产物(s,sp)
‑1‑
[4
‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基]
‑2‑
羟基二(二茂铁基)甲基二茂铁(1g)。
[0083]
产物结构式:
[0084]
产物性状为橙色固体,产率88%,熔点:116
‑
117℃,旋光度[α]
d20
=
‑
125.0(浓度0.34g/100ml,溶剂二氯甲烷);核磁氢谱(600mhz,溶剂氘代氯仿)δ8.65(s,1h,cfc2oh),4.79(s,1h,cp
‑
h),4.71(s,1h,cp
‑
h),4.61(s,1h,cp
‑
h),4.44(t,j=8.4hz,1h,chn),4.33(t,j=9.2hz,1h,ochh),4.27(s,1h,cp
‑
h),4.24
–
4.20(m,1h,ochh),4.17(s,1h,cp
‑
h),4.14(s,1h,cp
‑
h),4.12(d,j=3.6hz,2h,cp
‑
h
×
2),4.11
–
4.05(m,11h,cp
‑
h
×
11),3.99(s,1h,cp
‑
h),3.89(d,j=11.9hz,6h,cp
‑
h
×
5+cp
‑
h),1.52(s,3h,ch3),1.44(s,3h,ch3),0.22(s,9h,si(ch3)3).产物核磁碳谱(151mhz,溶剂氘代氯仿)δ169.46(c=n),100.14,76.32,74.54,73.36,72.08,70.81(cp
‑
c
×
5),69.98,69.38,68.70(cp
‑
c
×
5),68.48,67.84,66.78,66.28,65.94,65.03,28.97(ch3),25.18(ch3),2.62(si(ch3)3).高分辨质谱:(esi
‑
tof)计算值c
40
h
46
fe3no3si[m+h]
+
:784.1292;实际测量值784.1289。
[0085]
实施例12
[0086]
在高纯氮气保护下,在100ml干燥反应瓶中加入300mg(s)
‑4‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基二茂铁(5d,实施例4制得),使用高纯氮气置换三次;随后向反应体系中加入15ml无水无氧甲苯,将上述原料完全溶解;随后向反应体系中加入850mg新蒸二乙二醇二正丁醚。利用冰丙酮浴将反应体系降温至
‑
78℃后,向体系滴加1.0ml正丁基锂的正己烷溶液(1.6m),20分钟加毕;滴加完毕后,在
‑
78℃条件下继续反应2小时后,加入283mg重结
晶二苯甲酮;将体系缓慢恢复至室温,反应2小时;反应完毕,向体系中加入冰的饱和碳酸氢钠溶液;无水乙醚萃取反应液,直至水层无色;合并油相,无水硫酸镁干燥;将油相浓缩旋干溶剂,柱层析分离(洗脱剂石油醚:乙酸乙酯=18:1)得到产物(s,rp)
‑1‑
[4
‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基]
‑2‑
羟基二苯基甲基二茂铁(2)。
[0087]
产物结构式:
[0088]
产物性状为橙色固体,产率73%。非对应异构体分析条件:手性ad
‑
h柱,流动相:正己烷/异丙醇=99:1,1ml/min,保留时间:5.6min(s,sp构型);11.2min(s,rp构型);非对应异构体比例(s,sp):(s,rp)=6:1。高分辨质谱:(esi
‑
tof)计算值c
31
h
38
fen3o2si[m+h]
+
:568.2078;实际测量值568.2035。
[0089]
效果实验:
[0090]
实施例13
[0091]
将反应用schlenk反应瓶在110℃条件下烘干2小时,趁热接入无水无氧反应系统中。待反应瓶冷却后,在高纯氮气保护下,首先加入0.05mmol(s,sp)
‑1‑
[4
‑
(叔丁基二甲基硅氧基)甲基
‑
噁唑啉
‑2‑
基]
‑2‑
羟基二苯基甲基二茂铁(1a,实施例5制得),及2毫升新蒸无水无氧甲苯溶液,室温搅拌10分钟后,将反应体系降温至0℃。随后加入2ml 1.0m二乙基锌的正己烷溶液,在0℃搅拌30分钟后,利用注射器向反应体系中加入1mmol新蒸苯甲醛。将反应体系恢复到室温后,搅拌过夜。反应进度利用薄层色谱法进行检测。反应完毕后,向反应体系中加入4毫升饱和氯化铵溶液进行淬灭,并用3毫升乙醚洗涤三次。合并有机相,用饱和食盐水洗涤,无水硫酸镁干燥。粗产品柱层析分离,得到加成醇产物为无色油状液体。手性液相条件:手性od
‑
h柱,流动相:正己烷/异丙醇=99:1,1ml/min,保留时间:12.3min;15.0min。产率大于95%,ee=87%。
[0092]
实施例14
[0093]
将反应用schlenk反应瓶在110℃条件下烘干2小时,趁热接入无水无氧反应系统中。待反应瓶冷却后,在高纯氮气保护下,首先加入0.05mmol(s,sp)
‑1‑
[4
‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基]
‑2‑
羟基二苯基甲基二茂铁(1b)(实施例6制得),及2毫升新蒸无水无氧甲苯溶液,室温搅拌10分钟后,将反应体系降温至0℃。随后加入2ml 1.0m二乙基锌的正己烷溶液,在0℃搅拌30分钟后,利用注射器向反应体系中加入1mmol新蒸苯甲醛。将反应体系恢复到室温后,搅拌过夜。反应进度利用薄层色谱法进行检测。反应完毕后,向反应体系中加入4毫升饱和氯化铵溶液进行淬灭,并用3毫升乙醚洗涤三次。合并有机相,用饱和食盐水洗涤,无水硫酸镁干燥。粗产品柱层析分离,得到加成醇产物为无色油状液体,手性液相条件同实施例13。产率大于95%,ee=93%。
[0094]
实施例15
[0095]
将反应用schlenk反应瓶在110℃条件下烘干2小时,趁热接入无水无氧反应系统中。待反应瓶冷却后,在高纯氮气保护下,首先加入0.05mmol(s,sp)
‑1‑
[4
‑
(三甲基硅氧基二乙基)甲基
‑
噁唑啉
‑2‑
基]
‑2‑
(羟基二苯基)甲基二茂铁(1c,实施例7制得),及2毫升新蒸无水无氧甲苯溶液,室温搅拌10分钟后,将反应体系降温至0℃。随后加入2ml 1.0m二乙基
锌的正己烷溶液,在0℃搅拌30分钟后,利用注射器向反应体系中加入1mmol新蒸苯甲醛。将反应体系恢复到室温后,搅拌过夜。反应进度利用薄层色谱法进行检测。反应完毕后,向反应体系中加入4毫升饱和氯化铵溶液进行淬灭,并用3毫升乙醚洗涤三次。合并有机相,用饱和食盐水洗涤,无水硫酸镁干燥。粗产品柱层析分离,得到加成醇产物为无色油状液体,手性液相条件同实施例13。产率大于95%,ee=92%。
[0096]
实施例16
[0097]
将反应用schlenk反应瓶在110℃条件下烘干2小时,趁热接入无水无氧反应系统中。待反应瓶冷却后,在高纯氮气保护下,首先加入0.05mmol s,sp)
‑1‑
[4
‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基]
‑2‑
羟基二(二茂铁基)甲基二茂铁(1g,实施例11制得),及2毫升新蒸无水无氧甲苯溶液,室温搅拌10分钟后,将反应体系降温至0℃。随后加入2ml 1.0m二乙基锌的正己烷溶液,在0℃搅拌30分钟后,利用注射器向反应体系中加入1mmol新蒸苯甲醛。将反应体系恢复到室温后,搅拌过夜。反应进度利用薄层色谱法进行检测。反应完毕后,向反应体系中加入4毫升饱和氯化铵溶液进行淬灭,并用3毫升乙醚洗涤三次。合并有机相,用饱和食盐水洗涤,无水硫酸镁干燥。粗产品柱层析分离,得到加成醇产物为无色油状液体,手性液相条件同实施例13。产率大于95%,ee=93%。
[0098]
实施例17
[0099]
将反应用schlenk反应瓶在110℃条件下烘干2小时,趁热接入无水无氧反应系统中。待反应瓶冷却后,在高纯氮气保护下,首先加入0.05mmol(s,rp)
‑1‑
[4
‑
(三甲基硅氧基二甲基)甲基
‑
噁唑啉
‑2‑
基]
‑2‑
(羟基二苯基)甲基二茂铁(实施例12制得),及2毫升新蒸无水无氧甲苯溶液,室温搅拌10分钟后,将反应体系降温至0℃。随后加入2ml 1.0m二乙基锌的正己烷溶液,在0℃搅拌30分钟后,利用注射器向反应体系中加入1mmol新蒸苯甲醛。将反应体系恢复到室温后,搅拌过夜。反应进度利用薄层色谱法进行检测。反应完毕后,向反应体系中加入4毫升饱和氯化铵溶液进行淬灭,并用3毫升乙醚洗涤三次。合并有机相,用饱和食盐水洗涤,无水硫酸镁干燥。粗产品柱层析分离,得到加成醇产物为无色油状液体,手性液相条件同实施例13。产率大于95%,ee=
‑
12%。
[0100]
对照例1
[0101]
将反应用schlenk反应瓶在110℃条件下烘干2小时,趁热接入无水无氧反应系统中。待反应瓶冷却后,在高纯氮气保护下,首先加入2毫升新蒸无水无氧甲苯溶液,并将反应体系降温至0℃。随后加入2ml 1.0m二乙基锌的正己烷溶液,在0℃搅拌30分钟后,利用注射器向反应体系中加入101.5μl新蒸苯甲醛。将反应体系恢复到室温后,搅拌过夜。反应进度利用薄层色谱法进行检测。反应完毕后,向反应体系中加入4毫升饱和氯化铵溶液进行淬灭,并用3毫升乙醚洗涤三次。合并有机相,用饱和食盐水洗涤,无水硫酸镁干燥。粗产品柱层析分离,得到加成醇产物为无色油状液体。手性液相条件:正己烷/异丙醇=98:2,1ml/min,保留时间:14.2min;17.3min。ee=0%。
[0102]
对照例2
[0103]
将反应用schlenk反应瓶在110℃条件下烘干2小时,趁热接入无水无氧反应系统中。待反应瓶冷却后,在高纯氮气保护下,首先加入底物苯甲醛0.05倍当量的(r)
‑4‑
羟基甲基噁唑啉二茂铁,加入底物1.5倍量的二乙基锌。随后在0℃条件下加入新蒸苯甲醛,在该条件下反应24小时。产物1
‑
苯基丙醇产率大于95%,ee=36%。
[0104]
对照例3
[0105]
将反应用schlenk反应瓶在110℃条件下烘干2小时,趁热接入无水无氧反应系统中。待反应瓶冷却后,在高纯氮气保护下,首先加入底物苯甲醛0.1倍当量的(r)
‑4‑
(羟基二甲基)甲基噁唑啉二茂铁,加入底物2.2倍量的二乙基锌。随后在0℃条件下加入新蒸苯甲醛,在该条件下反应48小时。产物1
‑
苯基丙醇产率89%,ee=20%。
[0106]
对照例4
[0107]
将反应用schlenk反应瓶在110℃条件下烘干2小时,趁热接入无水无氧反应系统中。待反应瓶冷却后,在高纯氮气保护下,首先加入底物苯甲醛0.1倍当量的(s,sp)
‑
[5,5
‑
二甲基
‑4‑
异丙基噁唑啉]
‑2‑
(羟基二苯基)甲基二茂铁,在
‑
20℃条件加入底物2倍量的二乙基锌,反应20分钟。随后在该温度下加入新蒸苯甲醛,在该温度下反应24小时。产物1
‑
苯基丙醇产率32%,ee=33%。
[0108]
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,本领域普通技术人员对本发明的技术方案所做的其他修改或者等同替换,只要不脱离本发明技术方案的精神和范围,均应涵盖在本发明的权利要求范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1