一种氟吡菌酰胺的合成方法与流程
1.本发明涉及有机化学中间体的合成领域,具体涉及一种氟吡菌酰胺的合成方法。
背景技术:
2.氟吡菌酰胺(iso通用名:fluopyram),商品名称:路富达;cas no:658066
‑
35
‑
4,化学名称:n
‑
{2
‑
[3
‑
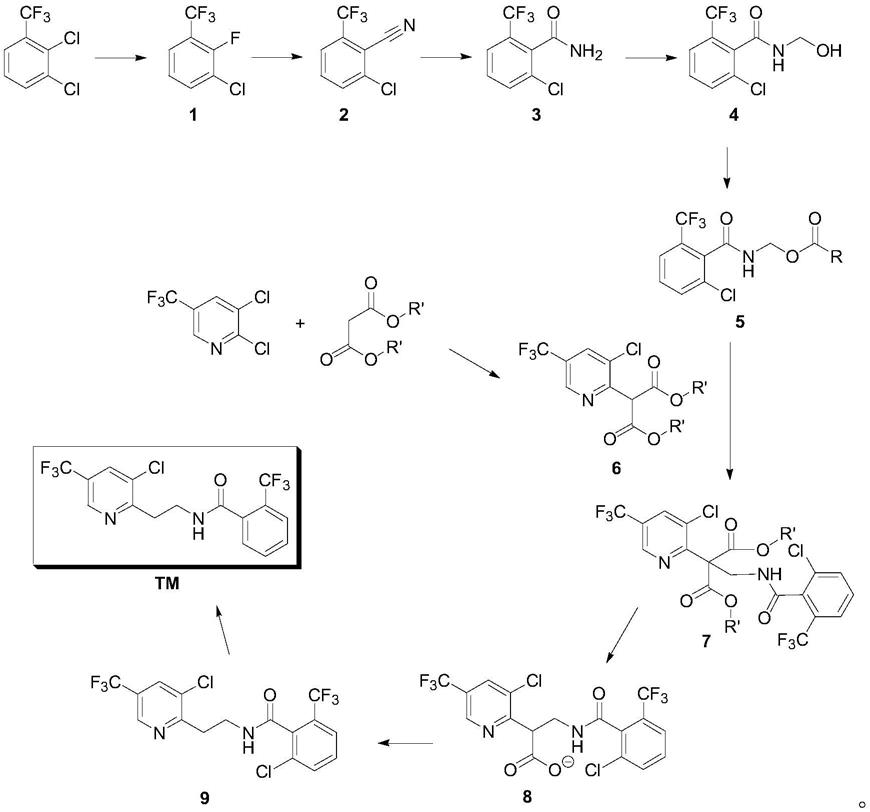
氯
‑5‑
(三氟甲基)
‑2‑
吡啶基]乙基}
‑
α,α,α
‑
邻三氟甲基苯甲酰胺,是拜耳作物科学公司开发的一种高效、绿色、低毒的sdhi(琥珀酸脱氢酶抑制剂)类杀菌、杀线虫剂。氟吡菌酰胺于2012年上市,迄今已在全球50多个国家登记和上市。主要用于大田作物、蔬菜、花卉、草坪、瓜果、烟草等70多种作物田的疫病防控。此外,还可作为广谱杀菌剂、种子处理剂、农产品仓储保鲜剂等,具有多功能性。
[0003]
德国拜耳作物科学公司2003年8月8日在中国申请化合物发明专利(专利号:038194716)。并于2012年11月5日在中国分别获得:96%氟吡菌酰胺原药农药登记(登记证号pd20121673)和41.7%氟吡菌酰胺制剂农药登记(登记证号pd20121664)。
[0004]
已公开的氟吡菌酰胺的合成主要有以下两种方法:
[0005]
方法一:专利ep1674455公开了以2,3
‑
二氯
‑5‑
三氟甲基吡啶和氰乙酸乙酯为原料,经过亲核取代、水解脱羧、催化氢化、水解制得3
‑
氯
‑5‑
(三氟甲基)
‑2‑
(2
‑
氨基乙基)吡啶,最后与中间体邻三氟甲基苯甲酰氯反应得到氟吡菌酰胺。
[0006][0007]
方法二:专利wo2018/114484公开了以邻三氟甲基苯甲酸为原料,经酰化、氨化、羟甲基化、酯化制成中间体b4;另以2,3
‑
二氯
‑5‑
三氟甲基吡啶与丙二酸二乙酯为原料缩合制成中间体b5,然后将中间体b4与中间体b5进行缩合制成中间体b6,最后水解脱羧得到氟吡菌酰胺。
[0008][0009]
通过比对可以发现,路线一虽然合成步骤少,但是中间体a3、a4的制备条件严苛,易生成多种杂质,难以纯化,使产品质量不易提高;路线二以邻三氟甲基苯甲酸为原料,成本高,且反应过程中释放大量酸性腐蚀性气体,对环境不友好。
[0010]
因此,开发一种条件温和、操作简单、成本低、三废少、收率高、纯度高氟吡菌酰胺的合成方法是亟待解决的问题。
技术实现要素:
[0011]
因此,本发明要解决的技术问题在于克服现有技术中的氟吡菌酰胺产率低、成本高、污染大的缺陷,从而提供一种条件温和、操作简单、三废少、收率高的氟吡菌酰胺的合成方法。
[0012]
为此,本发明提供了一种氟吡菌酰胺的合成方法,包括如下步骤:
[0013]
a)将2,3
‑
二氯三氟甲苯和氟化试剂溶解于第一溶剂中,加入第一催化剂,于
‑
20~260℃反应0.5~8h后,蒸馏得到中间体1;
[0014]
b)将中间体(1)加入于第二溶剂中,加入氰化试剂,于
‑
20~260℃反应2~8h后,得中间体2;
[0015]
c)将第二催化剂溶解于第三溶剂中,加入中间体(2),于
‑
20~260℃反应1~10h,得中间体3;
[0016]
d)将中间体(3)加入至第四溶剂中,加入第三催化剂,于
‑
20~260℃加入甲醛,反应1~16h,得中间体4;
[0017]
e)将中间体(4)溶解于第五溶剂中,加入第四催化剂,于
‑
20~260℃加入酰化试剂反应1~16h,得中间体5;
[0018]
f)将2,3
‑
二氯
‑5‑
三氟甲基吡啶加入第六溶剂中,加入第五催化剂,于
‑
20~260℃加入丙二酸二酯,反应1~16h,得中间体6;
[0019]
g)将中间体5加入到中间体6中,于
‑
20~260℃反应1~16h,得中间体7;
[0020]
h)将中间体7溶解于第七溶剂中,加入第六催化剂,于
‑
20~260℃水解反应1~16h,得中间体8;
[0021]
i)将第七催化剂加入到中间体8,于
‑
20~260℃反应1~16h,得到中间体9;
[0022]
j)将中间体9溶解于第八溶剂中,加入第八催化剂和第九催化剂,通入氢气,于1~10大气压,
‑
20~260℃反应1~16h,得到产物氟吡菌酰胺。
[0023]
反应过程为:
[0024][0025]
优选地,所述氟化试剂为氟化钠、氟化钾或氟化钙中的任意一种;所述第一催化剂为三苯基溴化磷、三苯基氯化磷、三苯基磷、二氮杂二环、三乙胺或二异丙基乙胺中的任意一种;所述第一溶剂为氯苯、二氯苯、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、二甲亚砜、n
‑
甲基吡咯烷酮、环丁砜或1,3
‑
二甲基咪唑啉酮中的任意一种;所述2,3
‑
二氯三氟甲苯、氟化试剂和第一催化剂的摩尔比为1:1.2~2.0:0.1~1.5。
[0026]
优选地,所述氰化试剂为氰化钠、氰化钾或氰化亚铜中的任意一种;所述第二溶剂为氯苯、二氯苯、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、二甲亚砜、n
‑
甲基吡咯烷酮、环丁砜或1,3
‑
二甲基咪唑啉酮中的任意一种;所述中间体1和氰化试剂的摩尔比为1:0.9~3.0。
[0027]
优选地,所述第二催化剂为氢氧化钠、氢氧化钾、氢氧化锂中的任意一种;所述中间体2和第二催化剂的摩尔比为1:0.1~5.0;所述第三溶剂为水、甲醇、乙醇中的任意一种或两种和两种以上的组合。
[0028]
优选地,所述第三催化剂为碳酸钾、碳酸钠、碳酸锂、碳酸铯中的任意一种;所述第四溶剂为水、甲醇、乙醇中的任意一种或两种和两种以上的组合;所述中间体3和第三催化剂的摩尔比为1:0.05~3.0。
[0029]
优选地,所述第四催化剂为三乙胺、二异丙基乙胺、三辛胺、二氮杂二环(dbu)中的任意一种;所述酰化试剂为乙酸酐、乙酰氯、丙酸酐、丙酰氯、对甲苯磺酰氯、甲磺酰氯中的任意一种;所述第五溶剂为二氯甲烷、二氯乙烷、乙酸乙酯、甲苯、二甲苯、氯苯、1,4
‑
二氧六
环、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、二甲基亚砜、n
‑
甲基吡咯烷酮、环丁砜或1,3
‑
二甲基咪唑啉酮中的任意一种;所述中间体4、第四催化剂和酰化试剂的摩尔比为1:0.9~3.0:0.9~3.0。
[0030]
优选地,所述第五催化剂为氢氧化钠、氢氧化钾、氢氧化锂、无水碳酸钾、无水碳酸钠、无水碳酸锂、无水碳酸铯、三乙胺、二异丙基乙胺、三辛胺、二氮杂二环(dbu)中的任意一种;所述第六溶剂为氯苯、二氯苯、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、二甲亚砜、n
‑
甲基吡咯烷酮、环丁砜或1,3
‑
二甲基咪唑啉酮中的任意一种;所述丙二酸二酯为丙二酸二甲酯、丙二酸二乙酯、丙二酸二丙酯中的任意一种;所述2,3
‑
二氯
‑5‑
三氟甲基吡啶、第五催化剂和丙二酸二乙酯的摩尔比为1:1.0~3.0:0.9~1.5;所述中间体5和中间体6的摩尔比为1:0.9~1.5。
[0031]
优选地,所述第六催化剂为氢氧化钠、氢氧化钾、氢氧化锂、无水碳酸钾、无水碳酸钠、无水碳酸锂、无水碳酸铯、三乙胺、二异丙基乙胺、三辛胺、二氮杂二环(dbu)中的任意一种;所述第七催化剂为盐酸、硫酸、磷酸、三氟乙酸、甲酸中的任意一种;所述第七溶剂为甲醇、乙醇、异丙醇、1,4
‑
二氧六环、乙醚、四氢呋喃、甲基叔丁基醚、水中的一种或两种或两种以上的组合;所述中间体7、第六催化剂和第七催化剂的摩尔比为1:0.9~3.5:0.9~5.0。
[0032]
优选地,所述第八催化剂为三乙胺、二异丙基乙胺、三辛胺、二氮杂二环(dbu)中的任意一种;所述第九催化剂为ranney镍、5%钯碳催化剂、10%钯碳催化剂、10%氢氧化钯碳催化剂、5%铂碳催化剂中的任意一种;所述第八溶剂为甲醇、乙醇、异丙醇、1,4
‑
二氧六环、乙醚、四氢呋喃、甲基叔丁基醚、水中的任意一种或两种或两种以上的组合。
[0033]
优选地,所述中间体8、第八催化剂和第九催化剂的摩尔比为1:0.9~3.5:0.05~0.5。
[0034]
本发明技术方案,具有如下优点:
[0035]
1.本发明提供的一种氟吡菌酰胺的合成方法,以2,3
‑
二氯三氟甲苯为原料,经过多步简单反应合成氟吡菌酰胺,各反应步骤中并未使用现有合成方法中常用的易燃易爆、剧毒或不易保存的试剂,避免对环境以及操作人员的危害;同时,产品的收率达到50%以上,纯度则在98%以上;
[0036]
2.本发明提供的一种氟吡菌酰胺的合成方法,具有各步反应操作简单,原料廉价易得,中间体1的异构体不参与后续反应,产品易纯化的特点;
[0037]
3.本发明提供的一种氟吡菌酰胺的合成方法,反应操作简单,产生的三废少,所用试剂容易回收,具有良好的工业化前景,提供了适合氟吡菌酰胺工业化大生产的新思路。
具体实施方式
[0038]
提供下述实施例是为了更好地进一步理解本发明,并不局限于所述最佳实施方式,不对本发明的内容和保护范围构成限制,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。
[0039]
实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。
[0040]
实施例1
[0041]
a)中间体1的制备
[0042][0043]
将215g 2,3
‑
二氯三氟甲苯、73g氟化钾、7g三苯基溴化磷和430ml 1,3
‑
二甲基咪唑啉酮加入装有精馏柱的1000ml三口瓶中,搅拌升温回流反应。反应1h后开始慢慢蒸出185g中间体1,为无色透明液体,gc分析含量93.9%。粗品收率93.2%。粗品不经纯化,直接用于下一步反应。
[0044]
b)中间体2的制备
[0045][0046]
180g中间体1溶解于360ml干燥n,n
‑
二甲基乙酰胺中,升温至90℃,分批慢慢加入42.0g氰化钠。加完后于90~100℃保温搅拌4小时,减压回收n,n
‑
二甲基乙酰胺。残余物溶解于900ml乙酸乙酯中,依次用水、饱和氯化钠水溶液洗涤,无水硫酸钠干燥,浓缩,残余物减压蒸馏,得到163.6g中间体2,为类白色固体。gc含量99.5%,收率87.6%
[0047]
c)中间体3的制备
[0048][0049]
24g氢氧化钠溶解于200ml水中,加入41.1g中间体2,升温到100℃搅拌反应4h,hplc分析原料反应完全。反应液降至室温,析出白色固体。抽滤,干燥,得到39.6g中间体3,hplc含量为96.8%,收率88.8%。
[0050]
d)中间体4的制备
[0051][0052]
22.4g中间体3与100ml水混合,加入1.0g碳酸钾,搅拌升温至90℃,慢慢滴加22ml37%甲醛水溶液,滴完加热回流反应16h,然后降至室温。反应液用二氯甲烷萃取,有机相水洗,无水硫酸钠干燥,减压浓缩,得到25.5g中间体4,为无色油状液体,hplc含量为92.4%,粗品收率100%。粗品不经纯化,直接用于下一步反应。
[0053]
e)中间体5的制备
[0054][0055]
25.5g中间体4、15g三乙胺与150ml二氯甲烷混合,室温搅拌下慢慢滴加11g乙酸酐,加完后于室温搅拌16h。反应结束后,反应液依次用水、饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩,得到29.9g中间体5,为无色油状液体,hplc含量为93.0%,粗品收率100%。粗品不经纯化,直接用于下一步反应。
[0056]
f)中间体6的制备
[0057][0058]
8.0g氢氧化钠与100ml n,n
‑
二甲基乙酰胺混合,搅拌升温至60℃,滴加22.0g 2,3
‑
二氯
‑5‑
三氟甲基吡啶与16.0g丙二酸二乙酯的混合溶液,滴完后保温搅拌6h,hplc检测原料反应完全,反应液直接用于下一步反应。
[0059]
g)中间体7的制备
[0060][0061]
29.9g中间体5加入到上一步得到的中间体6中,于65℃搅拌反应6h后,减压回收n,n
‑
二甲基乙酰胺。向残余物中加入150ml乙酸乙酯和100ml水,分液。有机相依次用水、饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩。粗品用乙醇重结晶,得到48.2g中间体7,为淡黄色固体。hplc含量为95.8%,收率:83.8%
[0062]
h)中间体8的制备
[0063][0064]
48.2g中间体7溶解于240ml95%乙醇中,加入100ml 3.0mol/l氢氧化钠溶液,反应液于60℃搅拌6h,得到澄清透明溶液,为中间体8的钠盐溶液。不经纯化,直接用于下一步反
应。
[0065]
i)中间体9的制备
[0066][0067]
然后向中间体8的钠盐溶液液中慢慢滴加30%浓盐酸,调ph至2.0,升温至60℃搅拌反应6h。减压蒸除溶剂,残余物加水搅拌,抽滤,粗品用乙醇重结晶,得到35.5g中间体9,为淡黄色固体,hplc含量为99.0%,收率:89.2%。1h
‑
nmr[dmso
‑
d6,300mhz]δ8.96(s,1h),8.79(br s,1h),8.42(s,1h),7.81(d,j=7.9hz,1h),7.73(d,j=7.7hz,1h),7.62(t,j=7.7hz,1h),3.90
‑
3.60(m,2h),3.50
‑
3.10(m,2h)。
[0068]
j)氟吡菌酰胺(tm)的制备
[0069][0070]
氮气保护下,35.5g中间体9溶解于100ml四氢呋喃中,加入15g三乙胺和3g5%钯碳催化剂,氢气置换,然后于室温、1.5atm氢气压力下搅拌至原料反应完全。反应液过滤,滤液减压浓缩,残余物加水稀释,析出白色固体,抽滤,滤饼水洗,干燥,得到30.2g氟吡菌酰胺tm,为类白色固体,hplc含量为98.6%,收率:92.4%。
[0071]
反应总产率为50.1%,产品hplc纯度为98.6%。
[0072]
实施例2
[0073]
a)中间体1的制备
[0074][0075]
将215g 2,3
‑
二氯三氟甲苯、83g氟化钠、10g二异丙基乙胺和400mln
‑
甲基吡咯烷酮加入装有精馏柱的1000ml三口瓶中,搅拌升温回流反应。反应4h后开始慢慢蒸出183g中间体1,为无色透明液体,gc分析含量93.5%。粗品收率92.2%。粗品不经纯化,直接用于下一步反应。
[0076]
b)中间体2的制备
[0077]
[0078]
180g中间体1溶解于360ml干燥二甲亚砜中,升温至90℃,分批慢慢加入177.1g氰化钾。加完后回流反应2小时,减压蒸馏后残余物溶解于900ml乙酸乙酯中,依次用水、饱和氯化钠水溶液洗涤,无水硫酸钠干燥,浓缩,残余物减压蒸馏,得到162.9g中间体2,为类白色固体。gc含量97.8%,收率87.2%。
[0079]
c)中间体3的制备
[0080][0081]
40gnaoh溶解于200ml水中,加入41.1g中间体2,升温到100℃搅拌反应4h,hplc分析原料反应完全。反应液降至室温,析出白色固体。抽滤,干燥,得到39.7g中间体3,hplc含量为96.9%,收率89.0%。
[0082]
d)中间体4的制备
[0083][0084]
22.4g中间体3与100ml水混合,加入3.2g碳酸钠,搅拌升温至90℃,慢慢滴加22ml37%甲醛水溶液,滴完加热回流反应16h,然后降至室温。反应液用二氯甲烷萃取,有机相水洗,无水硫酸钠干燥,减压浓缩,得到25.5g中间体4,为无色油状液体,hplc含量为92.5%,粗品收率100%。粗品不经纯化,直接用于下一步反应。
[0085]
e)中间体5的制备
[0086][0087]
25.5g中间体4、20g三乙胺与150ml二氯甲烷混合,室温搅拌下慢慢滴加20g丙酸酐,加完后于室温搅拌8h。反应结束后,反应液依次用水、饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩,得到29.7g中间体5,为无色油状液体,hplc含量为93.6%,粗品收率99.3%。粗品不经纯化,直接用于下一步反应。
[0088]
f)中间体6的制备
[0089]
[0090]
12.0g氢氧化钾与100ml二甲亚砜混合,搅拌升温至60℃,滴加22.0g 2,3
‑
二氯
‑5‑
三氟甲基吡啶与16.0g丙二酸二乙酯的混合溶液,滴完后保温搅拌16h,hplc检测原料反应完全,反应液直接用于下一步反应。
[0091]
g)中间体7的制备
[0092][0093]
29.7g中间体5加入到上一步得到的中间体6中,于70℃搅拌反应6h后,减压回收二甲亚砜。向残余物中加入150ml乙酸乙酯和100ml水,分液。有机相依次用水、饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩。粗品用乙醇重结晶,得到48.3g中间体7,为淡黄色固体。hplc含量为95.9%,收率:84.0%
[0094]
h)中间体8的制备
[0095][0096]
48.3g中间体7溶解于240ml95%乙醇中,加入100ml 3.0mol/l氢氧化钠溶液,反应液于80℃搅拌12h,得到澄清透明溶液,为中间体8的钠盐溶液。不经纯化,直接用于下一步反应。
[0097]
i)中间体9的制备
[0098][0099]
然后向中间体8的钠盐溶液液中慢慢滴加30%浓盐酸,调ph至2.0,升温至70℃搅拌反应6h。减压蒸除溶剂,残余物加水搅拌,抽滤,粗品用乙醇重结晶,得到35.8g中间体9,为淡黄色固体,hplc含量为99.1%,收率:90.0%。1h
‑
nmr[dmso
‑
d6,300mhz]δ8.96(s,1h),8.79(br s,1h),8.42(s,1h),7.81(d,j=7.9hz,1h),7.73(d,j=7.7hz,1h),7.62(t,j=7.7hz,1h),3.90
‑
3.60(m,2h),3.50
‑
3.10(m,2h)。
[0100]
j)氟吡菌酰胺(tm)的制备
[0101][0102]
氮气保护下,35.8g中间体9溶解于100ml甲醇中,加入10g三乙胺和2.5g5%钯碳催化剂,氢气置换,然后于室温、4atm氢气压力下搅拌至原料反应完全。反应液过滤,滤液减压浓缩,残余物加水稀释,析出白色固体,抽滤,滤饼水洗,干燥,得到30.6g氟吡菌酰胺tm,为类白色固体,hplc含量为98.7%,收率:93.6%。
[0103]
反应总产率为50.3%,产品hplc纯度为98.7%。
[0104]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1