一种噁唑烷酮类化合物的制备方法与流程
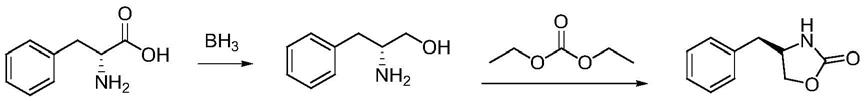
1.本发明属于医药中间体技术领域,具体涉及一种噁唑烷酮类化合物的制备方法。
背景技术:
2.噁唑烷酮类化合物在医药、有机合成和实际生产中有着广泛的用途,是具有极高应用价值的五元环化合物。首先,噁唑烷类化合物还在抑制细菌蛋白质合成反面具有极高的应用价值。其次,2
‑
噁唑烷酮及其衍生物是常用的手性辅助,其中4
‑
取代
‑2‑
噁唑烷酮作为代表具有很高的诱导选择性。evasn首先将其发展成广泛应用的手性辅助剂,即evans试剂。evans试剂已经有效应用在许多不对称的反应,diels
‑
alder反应和micheal反应、不对称羟醛缩合反应、羟基化反应等。因此不论以何种用的噁唑烷酮类化合物都有着极高的市场价值。其低成本的合成方法都显得更尤为重要。
3.早期噁唑烷酮的制备方法多以氨基醇为原料,用光气或光气类似物进行环化,此类有方法毒性大,实验操作繁琐,使用大量溶剂,浪费资源和污染环境等缺点。也有不少文献提出了不少与本专利类似的方法,但氨基醇作为重要的起始原料成本占比较高,且很多文献对于芳香族氨基醇的制备方法或生产成本较高或不适合大规模工业化生产;
4.如shengwei wei,regina messerer等用硼烷将氨基酸直接还原成氨基醇再经环化制备噁唑烷酮
[0005][0006]
以硼烷还原方式在反应过程中会生成氢气且过程温度变化较剧烈,在实际生产过程中较危险,硼烷在thf溶液中浓度低,因而造成物料成本和生产成本比较高,因而不宜工业化生产。
[0007]
makennon等发现nabh4‑
i2体系也能够直接还原氨基酸。α
‑
氨基酸被还原为相应的α
‑
氨基醇,产率在45%~94%;
[0008][0009]
当以此体系进行反应时,因产物水溶性较大会增加反应难度,导致收率较低。如,天冬酰胺酸和谷氨酸。
[0010]
wang x,li x,xue j等探索出了一个使用lialh4将酯还原成醇的方法,化学反应式。他发现lialh4/bncl还原体系具有很好的还原效果,用该体系在室温条件下对一系列酯进行还原,收率可以达到81%~94%。并且当lialh4为1.5倍当量时(酯∶lialh4∶bncl=1∶1.5∶1.5),产率最高。
[0011][0012]
虽然此反应收率较高,但锂铝氢价格昂贵且需要其他路易斯酸配合使用,导致在实际生产过程中物料成本占比较高。因此本专利提出一种制备噁唑烷酮类化合物的新工艺。
技术实现要素:
[0013]
本发明的目的在于:克服现有技术中的不足,提供一种反应温和、成本低、适用于工业化生产的噁唑烷酮类化合物的制备方法。
[0014]
为实现上述目的,本发明提供如下技术方案:
[0015]
一种噁唑烷酮类化合物的制备方法,所述制备方法包括以下步骤:
[0016]
1)酯化:将芳香族氨基酸溶于甲醇中,低温下投入氯化亚砜,升温到50
‑
60℃保温1
‑
2h,
[0017]
2)还原:以乙醇水为溶剂,硼氢化钠完全溶解后加入催化量的锂盐,再将酯化产品完全溶解于乙醇水溶液中,低温下缓慢滴加至硼氢化钠溶液中;
[0018]
3)关环:以甲苯为溶剂,投入还原产物及碳酸二乙脂升温至100℃,滴加甲醇钠溶液,滴加完毕后常压蒸馏至温度再升回100℃保温两小时经后处理及提纯后得到产品。
[0019]
进一步的,所述芳香族氨基酸的结构式为
[0020]
其中r=ph、bn。
[0021]
进一步的,所述步骤1)中芳香族氨基酸与氯化亚砜的摩尔比为1:1~1.2。
[0022]
进一步的,所述步骤1)中芳香族氨基酸与甲醇的质量比为1:3~7。
[0023]
进一步的,所述步骤2)中酯化产品与硼氢化钠的摩尔比为1:1~10。
[0024]
进一步的,所述步骤2)中酯化产品与硼氢化钠的摩尔比为1:2。
[0025]
进一步的,所述步骤2)中乙醇水溶液的质量分数为为30%~70%。
[0026]
进一步的,所述步骤2)中锂盐催化剂为氯化锂、溴化锂、碳酸锂中的一种,其与酯化产品的摩尔比为1:(0.01~0.1)。
[0027]
进一步的,所述步骤1)和步骤2)中的低温为0
‑
15℃。
[0028]
进一步的,所述步骤3)中甲苯的用量为还原产物质量的3~7倍。
[0029]
采用本发明中的技术方案的有益效果是:
[0030]
发明人在研发过程中发现锂盐在硼氢化钠还原过程中可有效减少空间位阻带来的影响,所以本发明中引入锂盐参与反应,但当锂盐不能游离在体系中形成自由离子,其对反应的优化作用并不能明显体现出来,因此本专利选择乙醇水为溶剂即可使硼氢化钠完全溶解,也可以将锂盐从化合物中游离出来以提高反应活性。使得硼氢化钠的使用量降低至2当量。对比硼氢化钠的其他体系其普遍用量为3~10当量,甚至更高,因此仅此一项就可显著降低生产成本。
[0031]
以(s)
‑4‑
苯基
‑2‑
噁唑烷酮为例,仅印度市场年需求大致为50t左右,当仅从3当量硼氢化钠降低至2当量时,每年生产成本可降低1000w元左右。因此本专利在降低生产成本上有较明显的优势。
具体实施方式
[0032]
下面将结合本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0033]
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
[0034]
下述实施例中所使用的材料及试剂等,如无特殊说明,均可从商业途径买到。
[0035]
一种噁唑烷酮类化合物的制备方法,所述制备方法包括以下步骤:
[0036]
1)酯化:将芳香族氨基酸溶于甲醇中,低温下投入氯化亚砜,升温到50
‑
60℃保温1
‑
2h,
[0037]
2)还原:以乙醇水为溶剂,硼氢化钠完全溶解后加入催化量的锂盐,再将酯化产品完全溶解于乙醇水溶液中,低温下缓慢滴加至硼氢化钠溶液中;
[0038]
3)关环:以甲苯为溶剂,投入还原产物及碳酸二乙脂升温至100℃,滴加甲醇钠溶液,滴加完毕后常压蒸馏至温度再升回100℃保温两小时经后处理及提纯后得到产品。
[0039][0040]
其中r=ph、bn
[0041]
研究发现锂盐在硼氢化钠还原过程中可有效减少空间位阻带来的影响,因此本专利引入锂盐参与反应,但当锂盐不能游离在体系中形成自由离子,其对反应的优化作用并不能明显体现出来,因此本专利选择乙醇水为溶剂即可使硼氢化钠完全溶解,也可以将锂盐从化合物中游离出来以提高反应活性。使得硼氢化钠的使用量降低至2当量。对比硼氢化钠的其他体系其普遍用量为3~10当量,甚至更高,因此仅此一项就可显著降低生产成本。
[0042]
以(s)
‑4‑
苯基
‑2‑
噁唑烷酮为例,仅印度市场年需求大致为50t左右,当仅从3当量硼氢化钠降低至2当量时,每年生产成本可降低1000w元左右。因此本专利在降低生产成本上有较明显的优势。
[0043]
在实际生产过程中,从经济性原则出发,我们优选了以下几个反应实例以供参考。
[0044]
实施例1:((s)
‑4‑
苄基
‑2‑
噁唑烷酮)
[0045]
1)酯化:在装有磁子的2l的三口烧瓶中,投入l
‑
苯丙氨酸250g、甲醇625g搅拌,冷却至0
‑
10℃,开始缓慢滴加197.6g氯化亚砜,过程控制温度不超过15℃,滴加完毕后,升温至50
‑
60℃,hplc检测原料≤0.5%进行后处理。
[0046]
后处理:冷却至室温后减压蒸馏至无溶剂蒸出后加入甲叔醚500g,升温至50~55℃搅拌0.5h,冷却至0
‑
5℃析晶2h,过滤50g甲叔醚淋洗两次再经干燥得类白色固体323.4g;
[0047]
2)还原:在装有机械搅拌的2l的三口烧瓶中,投入50%乙醇1000g,降温至0
‑
5℃加
入87.7g硼氢化钠、2.4g氯化锂开启搅拌使固体完全溶解。再将250g酯化产品完全溶解与500g 50%乙醇溶液中转移至滴液漏斗中,缓慢滴加过程控制温度不超过15℃,hplc检测原料≤0.5%进行后处理。
[0048]
后处理:反应液过滤后,滤饼用125ml无水乙醇淋洗两次后,旋干滤液,加入水250g,用dcm萃取四次,依次用1l、0.5l、0.5l和0.5l。合并有机相,并用盐碱水(187.5g水+62.5g氯化钠+4.6g氢氧化钠)洗涤一次后分液,有机相干燥旋干后的无色液体,再加入甲叔醚500g即析出大量白色固体冷却至0
‑
5℃析晶2h。过滤50g甲叔醚淋洗两次再经干燥得白色固体155.4g
[0049]
3)环化:在装有机械搅拌的1l的三口烧瓶中,投入甲苯650ml、还原产物130g和碳酸二甲酯121.9g升温至100℃,缓慢滴加46.4g30%甲醇钠溶液,滴加完毕后常压蒸馏至体系温度回到100℃撤去回流装置,保温2h,hplc检测原料≤0.5%进行后处理。
[0050]
后处理:降温至60℃,加入水260g,固体溶解后,分液,水相用130ml甲苯萃取一次,合并有机相,再加入20%氯化钠溶液260g加热至50
‑
55℃,分液,有机相冷却后减压蒸出溶剂析出大量固体,加入甲叔醚260g,0
‑
5℃析晶2h。过滤30g甲叔醚淋洗两次再经干燥得白色固体145.3g。
[0051]
实施例2:((s)
‑4‑
苯基
‑2‑
噁唑烷酮)
[0052]
1)酯化:在装有磁子的2l的三口烧瓶中,投入l
‑
苯甘氨酸250g、甲醇625g搅拌,冷却至0
‑
10℃,开始缓慢滴加216.8g氯化亚砜,过程控制温度不超过15℃,滴加完毕后,升温至50
‑
60℃,hplc检测原料≤0.5%进行后处理。
[0053]
后处理:冷却至室温后减压蒸馏至无溶剂蒸出后加入甲叔醚500g,升温至50~55℃搅拌0.5h,冷却至0
‑
5℃析晶2h,过滤50g甲叔醚淋洗两次再经干燥得类白色固体316.5g;
[0054]
2)还原:在装有机械搅拌的2l的三口烧瓶中,投入50%乙醇1000g,降温至0
‑
5℃加入94g硼氢化钠、2.7g氯化锂开启搅拌使固体完全溶解。再将250g酯化产品完全溶解与500g 50%乙醇溶液中转移至滴液漏斗中,缓慢滴加过程控制温度不超过15℃,hplc检测原料≤0.5%进行后处理。
[0055]
后处理:反应液过滤后,滤饼用125ml无水乙醇淋洗两次后,旋干滤液,加入水250g,用dcm萃取四次,依次用1l、0.5l、0.5l和0.5l。合并有机相,并用盐碱水(187.5g水+62.5g氯化钠+5.3g氢氧化钠)洗涤一次后分液,有机相干燥旋干后的无色液体,再加入甲叔醚500g即析出大量白色固体冷却至0
‑
5℃析晶2h。过滤50g甲叔醚淋洗两次再经干燥得白色固体162.4g
[0056]
3)环化:在装有机械搅拌的1l的三口烧瓶中,投入甲苯750ml、还原产物150g和碳酸二甲酯159.4g升温至100℃,缓慢滴加50g30%甲醇钠溶液,滴加完毕后常压蒸馏至体系温度回到100℃撤去回流装置,保温2h,hplc检测原料≤0.5%进行后处理。
[0057]
后处理:降温至60℃,加入水300g,固体溶解后,分液,水相用150ml甲苯萃取一次,合并有机相,再加入20%氯化钠溶液300g加热至50
‑
55℃,分液,有机相冷却后减压蒸出溶剂析出大量固体,加入甲叔醚300g,0
‑
5℃析晶2h。过滤50g甲叔醚淋洗两次再经干燥得白色固体165.6g。
[0058]
以上所述,仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专业的技术人
员,在不脱离本发明技术方案范围内,当可利用上述揭示的方法及技术内容作出些许的更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明技术方案的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1