一种核苷类化合物及其制备方法
1.本发明属于药物合成技术领域,尤其涉及核苷类化合物及其制备方法。
背景技术:
2.核苷类是具有广泛生理活性的一类水溶性成分。虫草素作为核苷类化合物,别名3'
‑
脱氧腺嘌呤核苷,化学式为c
10
h
13
n5o3。一直以来虫草素作为一种dna链延长的终止剂被广泛使用,尽管在很多细胞实验中,虫草素具有显著作用,但是其在进入体内后,大部分遵循嘌呤核苷代谢途径,在腺苷脱氨酶(ada)的作用下快速脱氨基而成为无活性的代谢产物“3'
‑
脱氧次黄嘌呤核苷”(agarwal rp,jr biochem pharmacol,1975,24(6):693
‑
701 &1977,26(5):359
‑
367),具体脱氨基反应如下所示:
[0003][0004]
fernandez
‑
noval a(leroy f.j endocrinology,1979,81(3):351
‑
354)等对受孕小鼠行卵巢切除术并用孕酮代替维持受孕状态的研究显示,虫草素进入体内尽管具有生物活性,但是作用短暂,并且需要较大剂量和较高浓度水平才可以实现。现有的解决方案中,包括虫草素侧链增加基团衍生物和采用虫草素与ada抑制剂联合应用,以达到延缓代谢,维持浓度的作用,但是虫草素侧链增加基团尽管能延缓代谢,但仍然不能完全阻断脱氨基作用,而使用ada抑制剂的使用虽然能抑制ada的活性,但是对于人体具有一定的副作用(例如引起严重的胃肠反应、骨髓毒性等),使用时对于虫草素的剂量浓度也有一定的影响,并且大大增加了用药成本。
[0005]
因此,尽管虫草素在体外实验中具有显著的活性作用,但虫草素的体内快速脱氨基代谢限制了药用价值的开发,现有的解决该缺陷的方案虽然在一定程度上可行,但仍然存在临床应用上的缺陷。
技术实现要素:
[0006]
本发明提供核苷类化合物及其制备方法,以解决现有技术中虫草素在体内遵循嘌呤核苷代谢途径,在腺苷脱氨酶的作用下快速脱氨基而成为无活性的代谢产物“3'
‑
脱氧次黄嘌呤核苷”从而快速失活的缺陷。
[0007]
为解决上述问题,本发明提供一种核苷类化合物,包括如下通式所示化合物,以及
药学上可接受的盐,其通式结构为:其中,r1代表r2=r3=ch3co 或h;或者,r1代表r2=r3=h。
[0008]
为解决上述问题,本技术还提供一种如上述所述核苷类化合物的制备方法,包括:以3'
‑
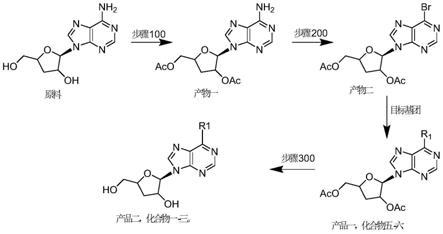
脱氧腺嘌呤核苷为原料,进行取代基上保护,得到产物一;对所述产物一,引入目标基团,得到产品一;或者,在所述步骤“对所述产物一,引入目标基团,得到产品一”之后,还包括:对所述产品一进行脱保护反应,得到产品二;其中,所述产品一或所述产品二为如上述所述的核苷类化合物。
[0009]
优选地,所述“以3'
‑
脱氧腺嘌呤核苷为原料,进行取代基上保护,得到产物一”包括:将原料3'
‑
脱氧腺嘌呤核苷与三乙胺和4
‑
二甲氨基吡啶混合,加入乙酸酐,得到混合物一;将所述混合物一加热至60℃,持续2小时;利用tlc跟踪反应,直至斑点消失,终止反应;将反应后的混合物干燥去除溶剂,剩余残渣重结晶纯化,即得到所述产物一;
[0010]
所述“对所述产物一,引入目标基团,得到产品一”包括:将所述产物一溶解于三溴甲烷中,加热至65℃;向该热溶液中加入亚硝酸叔丁酯,得到混合物二;将所述混合物二在65℃下搅拌2小时;利用tlc跟踪反应,直至斑点消失,终止反应;对终止反应物洗脱纯化,得到所述产物二;向所述产物二中加入目标基团化合物组,得到混合物二;利用tlc跟踪反应,直至原料斑点消失,终止反应;干燥去除溶剂,残渣余物洗涤纯化,得到所述产品一。
[0011]
优选地,所述目标基团化合物组为1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑
烯和对甲基苯硫酚、1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑
烯和苯酚,或者甲基苯胺;所述“向所述产品一中加入目标基团化合物组,得到混合物二”包括:向所述产品一中依次加入1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑
烯和对甲基苯硫酚,室温搅拌8小时,得到所述混合物二;或者,向所述产品一中依次加入1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑
烯和苯酚,室温搅拌8小时,得到所述混合物二;或者,将甲基苯胺加入至所述产品一中,65℃下加热8.5小时,即得到所述混合物二;
[0012]
所述“对所述产品一进行脱保护反应,得到产品二”包括:将氨水溶液添加至所述产品二中,室温搅拌0.5小时;利用tlc跟踪脱乙酰反应,直至斑点消失,终止反应;干燥并纯化,得到所述产品二。
[0013]
优选地,所述“以3'
‑
脱氧腺嘌呤核苷为原料,进行取代基上保护,得到产物一”包括:将原料3'
‑
脱氧腺嘌呤核苷与咪唑溶于dmf中;加入叔丁基二甲基氯硅烷,洗涤纯化后即得到所述产物一;所述“对所述产物一,引入目标基团,得到产品一”包括:将所述产品一溶解在吡啶中,加入2
‑
噻吩甲酰氯、呋喃甲酰氯或6
‑
氯烟酰氯进行反应,搅拌、冰浴、洗涤、干
燥,得到所述产品一;
[0014]
所述“对所述产品一进行脱保护反应,得到产品二”包括:将所述产品一溶于四氢呋喃,加入tbaf,室温搅拌反应;经减压干燥,硅胶柱纯化后得到所述产品二。
[0015]
此外,为解决上述问题,本技术还提供一种抗肿瘤药物,包含治疗有效量的选择上述所述核苷类化合物和/或药学上可接受的盐中的一种或多种作为活性成分,以及任选的药学上可接受的载体和/或赋形剂。
[0016]
此外,为解决上述问题,本技术还提供如上述核苷类化合物在制备用于作为抗癌的药物中的应用。
[0017]
此外,为解决上述问题,本技术还提供一种抗菌药物,包括有效量的作为活性成分的如上述所述核苷类化合物和药学上可以接受的载体。
[0018]
此外,为解决上述问题,本技术还提供一种如上述所述核苷类化合物在抗菌药用用品中的应用。
[0019]
优选地,包括应用于,一种可拆装白色念珠菌抑制口罩;所述可拆装白色念珠菌抑制口罩包括:佩戴主体,以及设于所述佩戴主体长度方向两端的双耳挂绳;所述佩戴主体包括外壳体和设于所述外壳体内侧且与所述外壳体可拆装连接的抑菌平面;所述抑菌平面上设有含有治疗有效量的所述核苷类化合物和/或其药学上可接受的盐中的一种或多种。
[0020]
本发明提供一种核苷类化合物及其制备方法。其中,所述核苷类化合物包括如下上述所示化合物,以及药学上可接受的盐。本发明提供一种核苷类化合物作为替代虫草素的衍生化合物,通过将6号位
‑
nh2取代基改变为芳香取代基,例如对甲基苯硫基、苯氧基和对甲基苯胺基,从而在体内能够有效避免嘌呤核苷代谢途径,保持药物剂量浓度充分发挥药物本身活性作用,避免产生药物在腺苷脱氨酶的作用下快速脱氨基而成为无活性的代谢产物“3'
‑
脱氧次黄嘌呤核苷”的缺陷。
附图说明
[0021]
图1为本技术中化合物一
‑
六的合成路线图示意图;
[0022]
图2为本技术中化合物七
‑
九的合成路线图示意图;
[0023]
图3为本技术中可拆装白色念珠菌抑制口罩正面及背面结构图;
[0024]
图4为本技术中可拆装白色念珠菌抑制口罩调整滑道局部放大图;
[0025]
图5为本技术中可拆装白色念珠菌抑制口罩的微针阵列组件剖面结构示意图;
[0026]
图6为本技术中可拆装白色念珠菌抑制口罩的微针阵列组件斜上方俯视,以及微针单体局部放大结构示意图;
[0027]
图7为本技术中可拆装白色念珠菌抑制口罩的抑菌滚动组件侧方位结构示意图;
[0028]
图8为本技术中可拆装白色念珠菌抑制口罩的抑菌滚动组件的斜上方俯视结构示意图;
[0029]
图9为本技术中可拆装白色念珠菌抑制口罩的滚动球的爆炸示意图;
[0030]
图10为本技术中可拆装白色念珠菌抑制口罩的滚动球剖面图,以及给药椎体局部放大示意图。
[0031]
图11为本技术中可拆装白色念珠菌抑制口罩的抑菌滚动组件在受到外力时发生形变的剖面结构及有效成分流向的示意图;
[0032]
图12为本技术中可拆装白色念珠菌抑制口罩的弹性渗漏管在接触人体皮肤并受到外力时结构及有效成分流向的示意图。
[0033]
本发明目的的实现、功能特点及优点将结合实施例,参照附图做进一步说明。
[0034]
附图标记:
[0035]
名称编号名称编号名称编号可拆装白色念珠菌抑制口罩1凝胶层1123半球托件1132佩戴主体11微阵列组件1124滚动球1133外壳体111微针单体1124a外凝胶球1133a外呼吸孔1111给药刺头1124a
‑
1内芯球1133b调整滑道1112给药管路1124a
‑
2给药椎体1133c抑菌平面112包合凝胶1124a
‑
3弹性渗漏管1133d内呼吸孔1121抑菌滚动组件113双耳挂绳12拆装基板1122基座1131
ꢀꢀ
具体实施方式
[0036]
下面结合具体实施例的方式对本发明的技术方案做进一步的详细说明,但并不构成对本发明的任何限制,任何人在本发明权利要求范围内所做的有限次的修改,仍在本发明的权利要求范围之内。
[0037]
本实施例中提供一种核苷类化合物,包括如下通式所示化合物,以及药学上可接受的盐,其通式结构为:其中,r1代表r2=r3=ch3co或h;或者, r1代表r2=r3=h。
[0038]
上述,根据通式中母核结构,本实施例中所提供的核苷类化合物为如下化合物:
[0039]
表1 化合物化学结构示意
[0040][0041]
本实施例提供一种核苷类化合物,包括如上表中所示化合物,以及其药学上可接受的盐。本实施例提供一种核苷类化合物作为替代虫草素的衍生化合物,通过将6号位
‑
nh2取代基替换为芳香取代基,例如对甲基苯硫基、苯氧基和对甲基苯胺基,从而在体内能够有效避免嘌呤核苷代谢途径,保持药物剂量浓度充分发挥药物本身活性作用,避免产生药物在腺苷脱氨酶的作用下快速脱氨基而成为无活性的代谢产物“3'
‑
脱氧次黄嘌呤核苷”的缺陷。
[0042]
此外,本实施例还提供一种核苷类化合物的制备方法,包括:步骤 100,以3'
‑
脱氧腺嘌呤核苷为原料,进行取代基上保护,得到产物一;步骤200,对所述产物一,引入目标基团,得到产品一;或者,在所述步骤200“对所述产物一,引入目标基团,得到产品一”之后,还包括:步骤 300,对所述产品一进行脱保护反应,得到产品二;其中,所述产品一或所述产品二为如上述所述的核苷类化合物。
[0043]
上述,3'
‑
脱氧腺嘌呤核苷即为原料药物
‑
虫草素。上述,步骤100为对于3'
‑
脱氧腺嘌呤核苷中2'号位
‑
oh和/或5'号位
‑
oh的保护,步骤200引入目标基团为对于6号位
‑
nh3取代基的取代反应引入相应的基团。步骤 300相应脱去步骤100对于2'号位
‑
oh和/或5'号位
‑
oh的保护,得到产品二。上述,产物一为反应过程中间体,产品一可以为表1中化合物4
‑
6、产品二可以为1
‑
3或6
‑
9中的任一化合物。
[0044]
进一步的,所述步骤100包括:步骤110,将原料3'
‑
脱氧腺嘌呤核苷与三乙胺和4
‑
二甲氨基吡啶混合,加入乙酸酐,得到混合物一;步骤 120,将所述混合物一加热至60℃,持续2小时;步骤130,利用tlc跟踪反应,直至斑点消失,终止反应;步骤140,将反应后的混合物干燥去除溶剂,剩余残渣重结晶纯化,即得到所述产物一;
[0045]
具体的,将3'
‑
脱氧腺嘌呤核苷(0.5g,1.99mmol)与三乙胺(1.19 ml,8.56mmol,4.3当量)和4
‑
二甲氨基吡啶(0.036g,0.298mmol, 0.15当量)在乙腈(20ml)中缓慢搅拌混合,然后缓慢加入乙酸酐(0.45 ml,4.78mmol,2.4当量)。将混合物一加热至60℃保持2小时,并且用 tlc跟踪反应进程,展开条件为甲醇:二氯甲烷=1:10,随时紫外下观察薄层板,直至原料斑点消失,则终止反应。利用真空干燥挥出有机溶剂,剩余固体残渣利用乙醇重结晶,即得到中间体产物一,2',5'
‑
二
‑
o
‑
乙酰基
ꢀ‑
3'
‑
脱氧腺苷。本实施例中,得到产物一为0.55g,产率83.6%,为白色固体。
[0046]
所述步骤200包括:步骤210,将所述产物一溶解于三溴甲烷中,加热至65℃;向该热溶液中加入亚硝酸叔丁酯,得到混合物二;步骤220,将所述混合物二在65℃下搅拌2小时;利用tlc跟踪反应,直至斑点消失,终止反应;对终止反应物洗脱纯化,得到所述产物二;步骤230,向所述产物二中加入目标基团化合物组,得到混合物二;利用tlc跟踪反应,直至原料斑点消失,终止反应;干燥去除溶剂,残渣余物洗涤纯化,得到所述产品一。
[0047]
将步骤140所得产物一(2',5'
‑
二
‑
o
‑
乙酰基
‑
3'
‑
脱氧腺苷,0.55g, 1.64mmol)溶于三溴甲烷(10ml)中,将其加热至65℃;向该热溶液中加入亚硝酸叔丁酯(3.9ml,32.8mmol,20当量)即得混合物二;将混合物在65℃下搅拌2小时。利用tlc跟踪反应(展开条件为甲醇:二氯甲烷=1:10,随时紫外下观察薄层板,直至原料点消失,终止反应)。终止反应后进一步洗脱纯化,得到产物二。具体的,终止反应物可以通过柱层析色谱法纯化,先用二氯甲烷洗脱,然后用2%甲醇/二氯甲烷洗脱,得到产物二,6
‑
溴嘌呤类似物。本实施例采用此方法得到产物二0.42g,产率 65.1%,呈粘稠黄色浆液的产物。向所述产物二中加入目标基团化合物组,得到混合物二;利用tlc跟踪反应,直至原料斑点消失,终止反应;干燥去除溶剂,残渣余物洗涤纯化,得到产品一。
[0048]
进一步的,所述目标基团化合物组为1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑ꢀ
烯和对甲基苯硫酚、1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑
烯和苯酚,或者甲基苯胺;所述步骤230,“向所述产品一中加入目标基团化合物组,得到混合物二”包括:步骤231,向所述产品一中依次加入1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑
烯和对甲基苯硫酚,室温搅拌8小时,得到所述混合物二;或者,步骤232,向所述产品一中依次加入1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑
烯和苯酚,室温搅拌8小时,得到所述混合物二;或者,步骤233,将甲基苯胺加入至所述产品一中,65℃下加热8.5小时,即得到所述混合物二。
[0049]
需要说明的是,目标基团化合物组,目的为引入衍生物目标取代基团,可以为单一化合物,也可以为多个化合物组合。具体的,可以包括但不限于:(1)1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑
烯和对甲基苯硫酚,对应步骤231;(2)1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑
烯和苯酚,对应步骤232,以及(3)甲基苯胺,对应步骤233。
[0050]
需要说明的是,步骤231
‑
233并不是连续的步骤,而是根据不同目标基团化合物组,引入不同取代基团为目的而择一进行。不同的目标基团化合物组,具体后续合成步骤上有所区别。
[0051]
上述,向产物二,即6
‑
溴嘌呤类似物(0.16g,0.4mmol)的无水乙腈 (8ml)溶液中加入目标基团化合物组。(1)在制备化合物1或4时,参考步骤231,向所述产品一中依次加入1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑
烯 (0.09ml,0.60mmol,1.5当量)和对甲基苯硫酚(0.25g,2mmol,5当量),室温搅拌8小时,得到所述混合物二;(2)在制备化合物2或5 时,参考步骤
232,向所述产品一中依次加入1,8
‑
二氮杂二环[5.4.0]十一碳
ꢀ‑7‑
烯(0.09ml,0.60mmol,1.5当量)和苯酚(0.19g,2mmol,5当量),室温搅拌8小时,得到所述混合物二;(3)在制备化合物3或6 时,参考步骤233,将甲基苯胺缓慢加入至所述产品一中(0.258g,2.4 mmol,6当量),65℃下加热8.5小时,即得到所述混合物二。
[0052]
利用紫外观察薄层板,直至原料斑点消失,终止反应。干燥去除溶剂,并将所得残余物溶于乙酸乙酯(30ml)。分别用5%氢氧化钠水溶液 (30ml)和盐水(30ml)洗涤有机层一次,无水硫酸镁干燥,过滤并减压浓缩。残余物经硅胶柱色谱纯化。
[0053]
需要说明的是,洗脱比例如果是在制备化合物1、2、4、5时则为甲醇:二氯甲烷=1:30;如果是在制备化合物3、6时则为甲醇:二氯甲烷=1:50;通过柱层析分离纯化得到产品一。其中,此步骤中可以得到产物为化合物4
‑
6。本实施例中,用上述方法得到化合物4为0.11g,产率 63.3%,为粘稠白色浆液;化合物5为0.09g,产率56.4%,为粘稠黄色浆液;化合物6为0.13g,产率74.3%,为粘稠黄色浆液。
[0054]
进一步的,所述步骤300包括:步骤310,将氨水溶液添加至所述产品二中,室温搅拌0.5小时;步骤320,利用tlc跟踪脱乙酰反应,直至斑点消失,终止反应;步骤330,干燥并纯化,得到所述产品二。
[0055]
上述,在制备化合物1时,将25%氨水(8ml)溶液添加到产品二 (经步骤311最终得到的化合物4,0.11g,0.25mmol;或者经步骤312 最终得到的化合物5,0.13g,0.32mmol;或者经步骤313最终得到的化合物6,0.13g,0.30mmol)的甲醇溶液(4ml)中,并将混合物在室温搅拌0.5小时后,利用tlc跟踪脱乙酰反应完成(展开条件:甲醇:二氯甲烷=1:9,紫外下观察薄层板,直至原料点消失,终止反应),干燥,并纯化即得到产品二。其中,纯化可以采用柱层析色谱(洗脱液比例为甲醇:二氯甲烷=1:9)分离纯化得到产品二,本实施例中,制备得到化合物1为0.06g,产率61.5%,为白色粉末;得化合物2为0.07g,产率 68.3%,为白色粉末;得到化合物2为0.07g,产率68.3%,为白色粉末。
[0056]
此外,在另一种实施方式中,针对化合物7
‑
9,所述步骤100包括:步骤150,将原料3'
‑
脱氧腺嘌呤核苷与咪唑溶于dmf中;步骤160,加入叔丁基二甲基氯硅烷,洗涤纯化后即得到所述产物一;
[0057]
本实施例中,将原料(1.5g,6mmol)和0.4446g咪唑(680mg,10 mmol)溶于6ml无水dmf(n,n
‑
二甲基甲酰胺)中;加入tbdmscl (叔丁基二甲基氯硅烷,1.5g,10mmol)搅拌5h后,真空除去溶剂,用乙酸乙酯66ml和水26ml萃取洗涤3次,有机相用naso4干燥浓缩得粗产物。将粗产物通过硅胶柱分离,甲醇
‑
二氯甲烷
‑
三乙胺(1:15)洗脱得到产物一,即5'
‑
o
‑
(叔丁基二甲基甲硅烷基)
‑6‑
噻吩甲酰胺
‑
3'
‑
脱氧腺苷。
[0058]
所述步骤200“对所述产物一,引入目标基团,得到产品一”包括:步骤240,将所述产品一溶解在吡啶中,加入2
‑
噻吩甲酰氯、呋喃甲酰氯或6
‑
氯烟酰氯进行反应,搅拌、冰浴、洗涤、干燥,得到所述产品一;
[0059]
上述,将产物一(0.5g,1mmol)溶解在干燥的吡啶(14ml)中,在n2保护下,向该溶液中加入2
‑
噻吩甲酰氯(针对化合物七,0.57g,4 mmol)、呋喃甲酰氯(针对化合物八,0.59g,4mmol)和6
‑
氯烟酰氯(针对化合物九,1.13g,4mmol)中的一种,室温下持续搅拌8h,将反应冷却至0℃,在冰浴中,缓慢加入nh4oh(28%aq)(4ml)。混合物搅拌 30分钟,在0℃下,真空除去溶剂,并将残余物溶于etoac(70ml),有机层用水,nahco3(水溶液)和盐水(35ml)洗涤,
并用na2so4干燥。将粗产物进行过柱子分离(meoh/ch2cl2,1:25)上硅胶柱进行分离,得到产品一。
[0060]
所述步骤300“对所述产品一进行脱保护反应,得到产品二”包括:步骤340,将所述产品一溶于四氢呋喃,加入tbaf(四丁基氟化铵水合物),室温搅拌反应;步骤350,经减压干燥,硅胶柱纯化后得到所述产品二。
[0061]
将产品一(0.6g,0.4mmol)的溶液溶于thf(四氢呋喃,13ml), tbaf(0.94ml,1.0m thf溶液)在0℃下加入,将反应体系在室温下搅拌5小时,然后真空浓缩去除溶剂;残留物通过硅胶柱纯化 (meoh/ch2cl2,1:20)得到产品二(化合物七、八、九)上硅胶柱进行分离,得到目标化合物,将产物进行干燥称重。
[0062]
表2 化合物结构表征(根据表1中化合物):
[0063]
[0064][0065][0066]
此外,本实施例还提供一种抗肿瘤药物,包含治疗有效量的选择核苷类化合物和/或药学上可接受的盐中的一种或多种作为活性成分,以及任选的药学上可接受的载体和/或赋形剂。此外,本实施例还提供一种核苷类化合物在制备用于作为抗癌的药物中的应用。
[0067]
上述,抗肿瘤抗癌所针对的为非实体瘤。所针对肿瘤细胞,为对肝癌细胞hepg2、乳腺癌细胞mcf7、胃癌细胞sgc
‑
7901。
[0068]
此外,本实施例还提供一种抗菌药物,其特征在于,包括有效量的作为活性成分的核苷类化合物和药学上可以接受的载体。
[0069]
此外,本实施例还提供一种核苷类化合物在抗菌药用用品中的应用。
[0070]
上述抗菌药用用品,为抗大肠杆菌、抗枯草杆菌、抗白葡萄球菌、抗白色念珠菌、抗金黄色葡萄球菌、抗绿脓杆菌中的一种或多种菌种的药用用品。上述抗菌药用用品包括口服制剂药物,也可以包括外用的药用用品等。
[0071]
进一步的,基于核苷类化合物在抗菌药用用品中的应用中,本实施例提供一种具体实施方式,针对于烧伤面部三角区域抗白色念珠菌应用的三角区抗菌口罩,包括应用于,一种可拆装白色念珠菌抑制口罩1。研究证明,严重烧伤创伤后,长期大量广谱抗生素致使耐药菌增加,常常导致肠道菌群失调,特别是肠道中的白色念珠菌,是系统性念珠菌感染(皮肤念珠菌病)的主要来源。而皮肤念珠菌病的病原体即为白色念珠菌,是一种由出牙培养的酵母样真菌。
[0072]
大多数念珠菌病可能是由导致疾病发生的内源性因素引起的。最常见情形是严重烧伤的患者机体免疫力下降,白色念珠菌突破免疫器官肠道的免疫屏障进而使身体其他部位发生感染。
[0073]
部分患者基于人体肠道菌群失调,致使白色念珠菌进入血液并感染后得皮肤念珠菌病,临床上主要有面部三角区的鹅口疮、面部皮肤上显示为红色、发炎、鳞状皮疹等表象。上述人体面部危险三角区之所以危险,是因为三角区血管丰富,炎症细菌极易扩散,更与颅底静脉相连,发生细菌感染后极易并发的引起颅底静脉炎。
[0074]
在用药时,白色念珠菌对一般抗生素不敏感,导致耐药菌株过度增殖,从而破坏了体内细菌群体之间的拮抗平衡。除上述外,外源性感染也不容忽视,即念珠菌病可因接触外界细菌而感染。
[0075]
由此可见,烧伤患者在感染白色念珠菌,并患有皮肤念珠菌病之后,如果需要暴露于室外进行移动,则会继续面临内源性和外源性的白色念珠菌感染的双重风险,现有的方法为内服抗生素,外出时采用普通口罩进行隔离,但并没有降低上述风险,治疗和预防效果较差。
[0076]
为解决上述问题,基于核苷类化合物在抗菌药用用品中的应用中,本实施例提供一种具体实施方式,包括:提供一种可拆装白色念珠菌抑制口罩1,针对于烧伤面部三角区域的皮肤念珠菌病。
[0077]
其结构所述可拆装白色念珠菌抑制口罩包括:佩戴主体11,以及设于所述佩戴主体11长度方向两端的双耳挂绳12;所述佩戴主体11包括外壳体111,以及设于所述外壳体111内侧,与所述外壳体111可拆装连接的抑菌平面112;所述抑菌平面112上设有含有治疗有效量的上述核苷类化合物和/或其药学上可接受的盐中的一种或多种作为活性成分,以及任选的药学上可接受的载体和/或赋形剂,能够经皮给药。
[0078]
进一步的,所述外壳体111为与人面部相适配的弧度结构,其外侧设有若干外呼吸孔1111,其内侧设有水平向设置的调整滑道1112;所述抑菌平面112设于所述调整滑道1112上,与所述外壳体111的弧度相适配,并且能沿所述调整滑道1112相对于所述内壳体移动;所述抑菌平面 112上设有能与所述外壳体111上的所述外呼吸孔1111相对应的内呼吸孔 1121,并且,内呼吸孔1121上设有过滤空气的喷绒布。
[0079]
上述,抑菌平面112,可以采用抑菌化合物对皮肤表面的贴附以达到对患者面部三角区所感染的白色念珠菌进行抑制,其平面整体为可更换结构,在患者佩戴一定时间后,可以经过拆卸,与外壳体111分离,丢弃后,更换新的抑菌平面112进行使用,从而达到有效避免二次感染的目的。
[0080]
进一步的,所述外壳体111面向所述抑菌平面112一侧的调整滑道 1112,设有至少两个,所述抑菌平面112能通过多个调整滑道1112相对于所述外壳体111沿着所述调整滑道1112长度方向移动。
[0081]
上述,患者的面部三角区局部,其感染部位或炎症部位可能并不完全置于面部三角区内,所以在佩戴时可能出现位置不对应,无法达到有效抑制体内、皮肤上的细菌菌、防护外界细菌感染等效果的情形。本实施例中,通过设置多个调整滑道1112进而能够在面部调整对应的位置。优选的,为上下两个横向设置的滑道,抑菌平面112卡进滑道后,能沿着滑道横向移动进行调整位置,以达到针对于特定区域、患处、创面的有针对性的保护和抑菌作用,与现有的其他口罩或贴片相比,能够更加灵活的针对患者患处进行调整,从而达到更好的效果。上述,抑菌平面112与调整滑道1112连接方式,可以为在抑菌平面112上设置t字滑块,而调整滑道 1112为设有滑动槽,t字滑块能置入所述滑动槽中,能沿槽横向滑动,此外也可以为其他连接方式,例如调整滑道1112为单轨,抑菌平面112设有滑轮,能卡设于该单轨上并通过滑轮往复移动。
[0082]
进一步的,具体实施方式1:所述抑菌平面112,包括有拆装基板1122,设于所述基板上的凝胶层1123,以及设于所述凝胶层1123背离所述拆装基板1122一侧的微阵列组件1124;所述微阵列组件1124包括设于其表面的等间距排列的多个微针单体1124a;所述微针单体1124a包括给药刺头1124a
‑
1,与所述给药刺头1124a
‑
1连接的给药管路1124
‑
2,以及包覆于所述给药管路1124
‑
2外围的包合凝胶1124
‑
3;所述给药刺头1124a
‑
1 和所述给药管路1124
‑
2中设有抑菌成分;
[0083]
上述,给药刺头1124a
‑
1能够刺破人体面部皮肤的角质层,并对皮肤角质层内进行给药,药物通过给药管路1124
‑
2、给药刺头1124a
‑
1进入皮肤内,以达到针对于白色念珠菌的抑菌效果。凝胶层1123和包合凝胶 1124
‑
3,与皮肤直接接触贴合,不会产生对皮肤的刺激感。
[0084]
抑菌平面112上微阵列组件1124,通过每个刺入皮肤的微针单体 1124a,组成给药的微阵列组件1124,对皮肤给药,从而达到对于白色念珠菌的抑菌效果,一方面,可拆装的抑菌平面112通过每一个微针单体 1124a刺入皮肤后产生固定效果,另一方面,利用本实施例中所提供的核苷类化合物达到对白色念珠菌的抑菌效果。此外,调整滑道1112能够更加有针对性的灵活的调整抑菌平面112的位置,使患处创面能够完全接触并达到给药位置,从而实现抑菌效果。总之,本实施例中所提供的可拆装白色念珠菌抑制口罩1,为核苷类化合物在制备抗菌药物中的具体应用方式,主要针对于烧伤患者的面部三角区域的皮肤念珠菌病,从而在烧伤患者短暂暴露于外界时,能够通过可拆装的抑菌平面112的微针阵列组件对患处进行实时给药,降低所面临的内源性和外源性的白色念珠菌感染的双重风险,为患者的烧伤创面的治疗提供了抑菌环境,同时口罩的佩戴也在一定程度上能够达到对于患者面部创面的遮挡作用,提高私密性,避免由于面部创面暴露给患者的外表造成影响和社交生活中带来的不便。
[0085]
此外,在另一种具体实施方式(具体实施方式2)中,面部三角区域烧伤患者,在佩戴用于抑菌的面罩口罩时,往往会由于长时间佩戴,面部创面与面罩相应药物层发生粘连、或者面罩与创面发生反复摩擦,由于摩擦力或者粘连导致创面再次破损导致二次感染,为患者皮肤细菌感染的治疗和康复造成较大影响。
[0086]
为解决上述问题,本实施例(具体实施方式2)中,抑菌平面112包括:拆装基板1122;所述拆装基板1122的一侧设有调整滑道1112,另一侧设有多个组成阵列的抑菌滚动组件113;每个所述抑菌滚动组件113包括与所述拆装基板1122连接的基座1131、设于所述基座1131上远离所述拆装基板1122一侧的半球托件1132,以及与所述半球托件1132卡接的滚动球1133;所述半球托件1132内设有一容置腔体,所述滚动球1133能置于所述半球托件1132的容置腔体内,以便于所述滚动球1133的裸露部分能与人体皮肤接触并基于所述半球托件1132的固定而在人体皮肤表面滚动。
[0087]
所述滚动球1133包括外凝胶球1133a,以及设于所述外凝胶球1133a 内的含有抑菌成分的内芯球1133b;所述内芯球1133b外表面设有给药椎体1133c;所述内芯球1133b内的抑菌成分能透过所述内芯球1133b外表面进入所述给药椎体1133c内;所述给药椎体1133c内设有弹性渗漏管 1133d,其一端与所述内芯球1133b外表面连通,另一端与所述外凝胶球 1133a连接,能将所述给药芯体内和给药椎体1133c内的抑菌成分在所述滚动球1133整体滚动时涂覆于人体皮肤表面。
[0088]
上述,外凝胶球1133a材质可以为凝胶材质,材质本身需要具有弹性,并且亲肤无刺激性的材质。内部内芯球1133b容置药物的有效成分,将有效成分包合在内芯球1133b内,给药椎体1133c可以为锥形体,其内可以设有与内芯球1133b内相同的,且能由内芯球1133b渗入有效成分的容置空间,给药椎体1133c与外凝胶球1133a之间隔离,防止药液渗出到外凝胶球1133a中。
[0089]
在所述外凝胶球1133a所受到外力发生变化时,对应位置的弹性渗漏管1133d能沿其长度方向发生弹性形变;当所述外凝胶球1133a受到外力后,所述弹性渗漏管1133d收缩,所述内芯球1133b内和所述给药椎体 1133c内的抑菌成分,能经所述弹性渗漏管1133d的管口和收缩状态的管身进入所述管内侧壁,并沿所述弹性渗漏管1133d穿过所述外凝胶球 1133a流道所述外凝胶球1133a表面。
[0090]
上述,给药椎体1133c内的弹性渗漏管1133d可以具有渗透性能,其本身材质可以为pp、橡胶、凝胶等材质,可以实现有效成分管内的渗入,优选的可以选择带有单向流动选择性透过的材质。弹性渗漏管1133d本身可以为弹簧状,在长度方向受到外力时,能够发生弹性形变,而给药椎体 1133c内的有效成分受到外力,更容易向弹性渗漏管1133d内渗入或注入药物,从而实现通过弹性渗漏管1133d对皮肤给药的功能。
[0091]
所述抑菌成分为上述产品二的三种化合物(化合物1
‑
3)中含有治疗有效量的核苷类化合物和/或药学上可接受的盐中的一种或多种作为活性成分,以及任选的药学上可接受的载体和/或赋形剂。
[0092]
优选的,所述抑菌成分为含有治疗有效量的化合物1或药学上可接受的盐中的一种或多种作为活性成分,以及任选的药学上可接受的载体和/或赋形剂;
[0093]
优选的,所述抑菌成分可以为聚合物
‑
化合物1偶联物的方式存在,例如,化合物一
与丁二酸二乙烯酯、已二酸二乙烯酯和/或葵二酸二乙烯酯聚合成为带有取代基(取代羟基)的聚合物
‑
核苷偶联物。以聚合物偶联物的形式存在,能够延长药物本身作用时间和毒副作用,大大提高缓释作用,从而提高可拆装白色念珠菌抑制口罩1的使用时间和抑菌效果。此外,为缓解患者面部烧伤的痛苦、提高皮肤修复能力,降低滚动球1133与皮肤接触的摩擦力,抑菌成分可以采用椰子油与产品二的三种化合物(化合物一
‑
三)混合,椰子油作为溶解载体,例如,可以为化合物一与椰子油0.05:10的配比的混合物,利用椰子油对于皮肤的修复功能,以及对于化合物的溶解性能,同时利用椰子油本身的润滑作用,涂敷在皮肤上降低对于皮肤的刺激性,抑菌同时修复皮肤。
[0094]
上述,在患者佩戴所述可拆装白色念珠菌抑制口罩1时,口罩内抑菌平面112上的滚动球1133与面部创面皮肤接触,即外凝胶球1133a与皮肤接触,外凝胶球1133a为亲肤凝胶成分,在与皮肤接触后能够在压力下发生形变,进而导致内部的弹性渗漏管1133d收缩,内芯球1133b内和给药椎体1133c内的抑菌成分就在收缩过程中流入到弹性渗漏管1133d内并流出内,通过滚动球1133外表面将抑菌成分涂覆于患者皮肤上,在佩戴过程中,由于面部动作或着走动过程中会发生口罩在患者面部的小范围移动,在移动过程中,滚动球1133外表面与皮肤一方面可以通过滚动摩擦大大降低摩擦力,减少患者不适感、疼痛感和二次感染的风险;另一方面通过滚动过程中,抑菌成分经弹性渗漏管1133d流出并在球体表面于患者皮肤处滚动涂覆,从而实现了药物对于患处在佩戴过程中的给药。
[0095]
综上,本实施例所提供的可拆装白色念珠菌抑制口罩1既可以通过口罩佩戴实现对于体外的细菌病毒的隔离,另一方面利用抑菌成分对于白色念珠菌的抑菌效果,对于来自体内进入皮肤的白色念珠菌实现有效抑制,同时,还能够在患者口罩使用过程中,利用口罩的小范围移动,实现自行给药的过程,为患者提供了方便,提供了更加安全的便利的给药途径。
[0096]
药理活性试验
[0097]
抗肿瘤实验:1、实验方法:(1)取hepg2、sgc
‑
7901和mcf7解冻。将细胞悬液添加10%的1640培养基调节细胞浓度,3
×
104个/ml计数并培养。(2)将虫草素,化合物一
‑
九制备成浓度为80、40、20、10、 5、2.5μmol/l溶液,以羟基喜树碱(hcpt)为阳性对照。将细胞以3
×
105个/ml接种量接种于培养板,培养24h后吸弃培养液,每孔加入不同浓度化合物、hcpt,恒温培养48h后吸弃上清。(3)加入100μl mtt后培养4h弃去培养液,每孔加入150μl dmso,在490nm下根据测得的吸光度值计算细胞生长抑制率,并计算各给药组抑制率以及半数抑制浓度 (ic
50
)。
[0098]
2、实验结果:
[0099]
表3、化合物对人体肿瘤细胞体外增殖抑制活性ic
50
值(μmol/l)
[0100]
试样mcf7hepg2sgc
‑
7901试样mcf7hepg2sgc
‑
7901虫草素46.8551.8351.27六767.56611.22740.67一157.96194.71162.76七27.5668.7938.92二312.18201.95137.52八40.9333.3686.30三147.2546.3476.65九45.2648.3680.39
四544.69853.46606.58hcpt8.566.567.96五548.92459.45866.89
ꢀꢀꢀꢀ
[0101]
综上,肿瘤抑制率实验中,以虫草素为对照、hcpt阳性对照,测定化合物对hepg2、mcf7和sgc
‑
7901的增殖抑制率。通过酶标仪测定 od值,将mtt实验结果进行处理后,计算出化合物对三种癌细胞的抑制率。如上表,化合物一
‑
九对于3种癌细胞均有不同程度活性抑制作用,其中,化合物一修饰基团为碱基上连有对甲基苯硫酚作为取代基时,对乳腺癌细胞和胃癌细胞有一定抑制作用,而对肝癌细胞无明显抑制作用;化合物三对肝癌细胞和胃癌细胞有明显的抑制作用,对乳腺癌细胞也有一定的抑制作用。与虫草素相比化合物七、八和九针对mcf7,八和九针对 hepg2,七针对sgc
‑
7901均有较强抑制率。
[0102]
抗菌试验:1、实验方法:(1)制备液体培养基和固体培养基;
[0103]
(2)活化菌株;(3)分装培养基,并配制菌液。刮取适量的菌落溶于灭菌生理盐水中备用。(4)最小抑菌浓度(mic)测定:将化合物稀释过滤,稀释使药物初始浓度为40mmol/l。将菌落溶于液体培养基培养中待用。六种菌液浓度均为1
×
108cfu/ml。将100μl的药物溶液分别加入板中。在给药孔和对照中加入菌液,恒温培养24h判断结果,通过酶标仪测定。
[0104]
2、实验结果:
[0105]
表4、化合物对6种菌的mic参考值(mmol/l)
[0106]
试样大肠杆菌枯草杆菌白葡萄球菌白色念珠菌金黄色葡萄球菌绿脓杆菌虫草素
‑
160160
‑
160
‑
一2.502.501.251.252.505.00二4080160808040三801601601608080四160160160808040五808080160160160六16080160160160160七80160160808080八1601601601608080九3015303031.257.5
[0107]
综上,对核苷类似物进行抗细菌活性检测,结果表明:虫草素对六种致病菌抑制作用不明显,化合物对于6种菌株均有不同程度的抑菌活性,其中,化合物一对大肠杆菌、枯草杆菌、白色念珠菌、金黄色葡萄球菌有较明显抑制作用,尤其是对于白色念珠菌和白葡萄球菌,其抑菌作用优于单独使用虫草素溶液的效果。化合物一修饰基团为碱基上连有对甲基苯硫酚作为取代基时对6个菌均有较明显的抑制作用。化合物九对六种致病菌均有明显抑制作用。
[0108]
可拆装白色念珠菌抑制口罩的皮肤抑菌试验和皮肤创面刺激性实验:对于可拆装白色念珠菌抑制口罩的两种实施方案,分别进行了皮肤抑菌实验,以及皮肤创面刺激性实验。
[0109]
皮肤抑菌现场实验:表5分组试样表
[0110]
试样口罩形式抑菌成分试样口罩形式抑菌成分1实施方式1,微阵列组件化合物一3实施方式2,抑菌滚动组件化合物九
2实施方式1,微阵列组件化合物一4实施方式2,抑菌滚动组件化合物九
[0111]
选定30名具有白色念珠菌感染患者,对每个患者的患处均进行如下操作并取样:(1)用无菌棉拭子沾湿采样液在皮肤创面5cm
×
5cm范围内往返擦涂2遍,作为空白对照组;(2)取该试样对应的口罩贴合于不同患处创面5分钟,用无菌棉拭子按照上述方法采样,分别获取试样1
‑ꢀ
4。取空白对照组、1
‑
4样品,经过混匀振荡洗脱稀释,分别接种无菌平皿,加入营养琼脂培养基37℃培养48h后观察结果。
[0112]
实验结果:经过对于30名志愿者皮肤抑菌实验证明,实验前患者创面白色念珠菌数为453cfu/cm2,经利用可拆装白色念珠菌抑制口罩(试样 1
‑
4)对患处实施贴附作用5分钟后,试样1对应白色念珠菌下降到均值 88cfu/cm2,除菌率为平均80.6%;试样2均值95cfu/cm2,除菌率为平均 78.9%;试样3对应白色念珠菌下降到均值67cfu/cm2,除菌率为平均 85.6%,试样3对应白色念珠菌下降到均值65cfu/cm2,除菌率为平均 87.6%,由此可见试样1
‑
4均具有较好的抑菌效果。
[0113]
皮肤创面刺激性实验:用健康家兔12只(四组,每组3只),试验前 24h将背部脊柱两侧毛分别去掉3cm
×
3cm,不损伤表皮。将该试样1
‑
4 分别固定在每只家兔右侧皮肤上,左侧皮肤用10%甘油作对照,敷用时间为12h。试验结束后用温水清洗,并于除受试物后1、24和48h观察皮肤反应,按照2002年版《消毒技术规范》中2.3.3.4.1评价刺激强度。
[0114]
实验结果:结果表明以本实施例中试样1
‑
4作为受试物,接触家兔皮肤后未见异常,24h后观察勉强可见红斑形成,48h观察无明显变化。对皮肤刺激反应平均积分值为1.91,对照一侧皮肤未见异常反应,说明试样1
‑
4对家兔皮肤并无强轻刺激性。
[0115]
以上所述的是本发明的优选实施方式和相应实施例,应当指出,对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提,还可以做出若干变形和改进,包括但不限于比例、流程、用量的调整,这些都属于本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1