一种液相捕获文库构建方法与流程
1.本发明属于分子生物学技术领域,具体涉及一种液相捕获文库构建方法。
背景技术:
2.精准医学是指根据每一位患者的特点来调整医学治疗措施的新型医学概念和医疗模式。精准医学的实现离不开高通量测序技术的支持。高通量测序技术(ngs)是一次对几十万到几百万条dna分子进行序列测定,是传统测序技术的一次革命性改变。高通量测序技术(ngs)的应用十分广泛。在无创产前诊断、肿瘤临床诊断、遗传性疾病检测等众多实践中都发挥着积极的作用。虽然高通量测序技术发展迅猛,但目前来看测序价格仍然是一个阻碍高通量测序在临床上发展应用的一个瓶颈。因此,目标区域捕获技术便应运而生。而基于探针的液相捕获方法是目前目标区域捕获技术的主流。因此基于探针的液相捕获技术其成本、效能便成为相关临床或检测工作者们广为关注的重点。目前基于探针的液相捕获技术和产品主要有罗氏的nimblegen、安捷伦的sureselect,以及twist bioscience的捕获产品等。它们普遍都是采用类似的方案,我们称之为传统技术方案(图1)。传统技术方案操作流程繁琐耗时(操作时间目前的商业化方案最短要12小时以上),成本高昂(传统技术方案需要使用大量的昂贵的接头封闭寡核苷酸),已经不能够满足日益增长的高通量测序检测需求。因此,亟待发展一种简化流程、成本更加低廉的探针液相捕获文库构建技术。
技术实现要素:
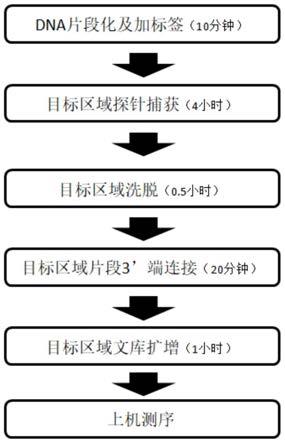
3.本发明的目的是提供一种有别于传统方法的dna探针液相捕获文库构建方法,详细技术流程见图2。
4.本发明的技术方案包括如下步骤:
5.一种液相捕获文库构建方法,包括以下步骤:
6.(1)获取进行了5’端接头连接的双链dna片段,双链dna片段的长度为100
‑
300bp;
7.(2)加入探针到步骤(1)的体系中捕获目标区域;
8.(3)捕获到的目标区域dna单链或者cdna,经过5’端磷酸化和3’端羟基化处理,进行3’端辅助连接,延伸成dna双链;
9.(4)对于步骤(3)获得的延伸产物采用通用引物直接进行扩增,得到目标区域文库。
10.上述的方法,对于多个样本在同一个探针体系中进行杂交反应,则需要在5’端加上含标签的接头,包括用来区分不同样本的标签,或者用来区分不同分子的标签,或者两者都含有。
11.上述的方法,对于长链双链dna分子使用tn5转座酶进行片段化并且在5’端添加接头,或者含标签的接头。经过tn5转座酶片段化后的dna片段不需要5’端磷酸化,其3’端天然已经是羟基。
12.上述的方法,5’端接头的长度范围19
‑
100bp,优选19
‑
90bp,进一步优选19
‑
80bp。
使用tn5转座酶进行片段化时,5’端接头必须至少要含有tn5转座酶的识别核心序列,其余序列只能加在该识别核心序列的5’端。
13.本发明对于长链双链dna分子使用tn5转座酶进行片段化并且添加分子标签(umi),或者添加用于区分不同样本的样本标签(index)。也可以将分子标签(umi)和样本标签(index)同时添加到dna的5’端。原理见图3。如果双链dna材料已经是100bp
‑
300bp左右的片段化dna,则可以使用传统方法进行5’端接头连接或含标签接头连接,以及5’端磷酸化和3’端羟基化处理。
14.本发明对于已经是碎片化的dna样本可以不需要进行上一步dna片段化并加标签,而是直接进行此目标区域探针捕获步骤。对于想要进行多个样本在同一个探针体系中进行杂交反应来降低成本的方案则需要进行上一步的dna片段化并加标签,此时加的标签可以是用来区分不同样本的index,也可以是用来区分不同分子的标签umi。也可以两者都含有。传统目标区域捕获方法必须要使用封闭寡核苷酸来封闭预文库的接头序列(图4),如果不封闭或者封闭效果不好,那么探针将有可能捕获到这些未被封闭掉的接头序列,同时已经被正确捕获到的预文库分子由于也含有通用的接头序列,这些接头序列也极有可能会去捕获到这些未被封闭掉的非目标区域的分子的接头序列。从而最终造成的结果是捕获效率低下。
15.本发明(图5)区别于传统目标区域捕获的方法的要点在于,本发明直接对样本进行目标区域捕获,不需要像传统目标区域捕获方法那样使用高浓度的封闭寡核苷酸来对总长达120bp左右的接头序列进行封闭占位。即便采用本发明中上述转座酶加标签的方案,由于本发明中使用了优化至只有十几个碱基的5’端接头,因此即便不使用封闭寡核苷酸进行封闭占位也能够得到很高的捕获效率。这样就实现了比传统方法更经济高效的目的。
16.本发明的上述方法步骤(3)对于捕获到的目标区域单链dna片段进行3’端辅助连接,具体构建流程如图6:
17.体系中使用一种人工合成的核苷酸片段辅助子,以及dntp,且dntp是普通的dntp和热启动dntp的混合物,普通dntp选用四种dntp中的任一种、两种或者三种,热启动的dntp为选用的普通dntp对应剩余的三种、两种或者一种;在较低温度条件下利用所述的普通dntp进行dna片段的加尾反应;然后给体系一个较高的温度,使热启动的dntp进入到激活工作的状态;再给一个相对较低的温度,使辅助子与dna片段3’端加尾序列互补结合,最后由dna聚合酶分别以辅助子和dna片段为模板向前延伸,直至以辅助子为模板延伸出辅助子的互补链,并且以dna片段为模板延伸出dna片段的互补链,辅助子3’端含有与dna片段加尾互补的序列,且3’末端需要进行封闭,以防止体系中的加尾酶对辅助子进行3’加尾,延伸时解除3’末端的封闭。
18.步骤(3)中:辅助子长度15nt
‑
120nt,优选30nt
‑
60nt,辅助子3’端至少第1个碱基采用rna碱基进行封闭;所述的rna碱基为ra、rg、rc或ru。
19.步骤(3)中:dna片段3’端加尾序列长度范围1
‑
20个碱基,优选3
‑
10个碱基,辅助子3’端与dna片段加尾序列互补结合的碱基序列在辅助子3’端rna碱基之后直到第14个碱基中的任意部分。
20.进一步地,通过限制普通dntp的量和/或增加辅助子的量来控制dna片段3’端加尾碱基数目,随着普通dntp的浓度降低,则dna片段3’端加尾碱基数目会减少;随着辅助子的
浓度增加,则dna片段3’端加尾碱基数目会减少。
21.进一步地,体系中辅助子浓度为10nm
‑
20um之间,优选50nm
‑
10um之间,进一步优选100nm
‑
5um。
22.进一步地,反应体系中捕获到的目标区域dna单链或者cdna浓度1fmol
‑
1nmol/50ul。
23.进一步地,体系中普通dntp的浓度为2um
‑
2mm,优选10um
‑
1mm之间,进一步优选30um
‑
0.5mm之间;体系中普通dntp浓度与每一种热启动dntp的1倍
‑
3倍。
24.步骤(3)中:首先在20℃
‑
37℃条件下进行dna片段的加尾反应5
‑
30分钟;然后在90℃
‑
95℃反应1
‑
5分钟,使热启动的dntp进入到激活工作的状态,同时使辅助子3’端的rna碱基从辅助子上掉下来,激活辅助子的延伸功能;再在45℃
‑
72℃反应2
‑
30分钟,使辅助子与dna片段3’端加尾序列互补结合,之后由dna聚合酶分别以辅助子和dna片段为模板向前延伸,直至以辅助子为模板延伸出辅助子的互补链,并且以dna片段为模板延伸出dna片段的互补链。
25.进一步地,体系中对于dna片段加尾实现方式,包括:使用加尾酶,以体系中加入的普通datp、dttp、dgtp和dctp,4种dntp中的任一种、两种或三种为材料进行dna片段的3’端加尾;
26.优选:体系中加尾酶的浓度为0.15u/ul
‑
15u/ul,优选0.4u/ul
‑
5u/ul,进一步优选0.6u/ul
‑
3u/ul。
27.进一步地,体系中用于聚合的聚合酶包括taq聚合酶中的至少一种,或者高保真聚合酶中的至少一种;taq聚合酶包括:2g robust dna聚合酶、rtaq dna聚合酶、taqb dna聚合酶中的一种或几种,高保真聚合酶包括:kapa hifi热启动高保真聚合酶、pfu dna聚合酶、phusion dna聚合酶中的一种或几种;优选taq dna聚合酶;
28.优选:体系中聚合酶的浓度为0.01u/ul
‑
2u/ul;优选0.05u/ul
‑
1u/ul,进一步优选0.08u/ul
‑
0.5u/ul。
29.本发明对于连接产物采用通用引物直接进行扩增,产生合适浓度的目标区域文库。见图7。
30.本发明同传统技术方法相比,省掉了构建预文库的步骤,整个文库构建时间从多于12小时缩短到了6小时以内,见图1、图2。并且在关键流程中创造性地省掉了传统技术方案中必须要使用的接头序列封闭寡核苷酸。这样,不仅使操作方法更简单,流程更简洁不易出错,而且极大降低了检测成本。在传统的液相捕获中,接头block寡核苷酸的成本甚至要占到整个液相捕获成本的一半以上。因此,本发明中创造性的避免了接头block寡核苷酸的使用,因此降低了检测成本。具有很好的应用前景。
附图说明
31.图1传统液相捕获方法流程及耗时示意图;
32.图2本发明液相捕获方法流程及耗时示意图;
33.图3本发明对长链双链dna分子通过tn5转座酶进行打断并添加标签序列原理图;
34.图4传统液相捕获方法原理图;
35.图5本发明液相捕获方法原理图;
36.图6本发明dna3’端辅助生成原理图;
37.图7本发明扩增终文库原理图;
38.图8本发明实施例1的电泳结果图;
39.图9本发明实施例2的电泳结果图;
40.图10本发明实施例3的电泳结果图;
41.图11本发明实施例4的电泳结果图;
42.图12本发明实施例5的电泳结果图。
具体实施方式
43.以下结合实施例旨在进一步说明本发明,而非限制本发明。
44.实施例1
45.单样本无标签目标区域文库构建
46.取人白细胞基因组dna 200ng,使用本发明方法(s1)构建目标区域文库。
47.1.dna片段化
48.转座酶识别核心序列:
49.tup1:
50.agatgtgtataagagacag,序列见seq id no.1,
51.tup2:
52.ctgtctcttatacacatct,序列见seq id no.2,
53.将tup1和tup2按等摩尔比进行退火,形成双链dna接头tup。
54.将转座酶和双链dna接头tup按如下流程组装在一起形成转座酶复合物备用。
55.转座酶复合物的组装:
56.首先将tup1和tup2等摩尔量在0.5m氯化钠溶液中进行退火生成双链产物tup。
57.配制如下反应体系:
58.tup(50um)4ultn5转座酶(1u/ul)5ul10x tps buffer1ul总体积10ul
59.pcr仪上25℃70min,4℃保存。转座酶复合物即组装完成。
60.dna片段化反应体系如下:
61.dna200ng转座酶复合物2ul5x lm buffer2ul去离子水补至10ul
62.pcr仪上55℃10min,4℃保存。
63.2x ampure xp beads纯化。
64.2.目标区域探针捕获
65.配制反应体系如下:
66.片段化dna20ulcot
‑
1 dna5ug
67.将上述反应体系在真空旋转蒸发仪中蒸干液体。
68.加入8.5ul nimblegen 2x杂交缓冲液,3.4ul nimblegen hybridization component a,1.1ul无酶水
69.配制反应体系如下:
70.2x杂交缓冲液8.5ulnimblegen hybridization component a3.4ul去离子水1.1ul
71.使用配制好的13ul液体体系重悬蒸干后的dna。转移到0.2ml pcr管中。
72.pcr仪上95℃10分钟。
73.加入4ul xgen pan
‑
cancer panel探针(idt:integrated
74.dna technoligies#1056205),混匀后瞬时离心。
75.pcr仪上65℃杂交4小时。
76.3.漂洗
77.取100ul dynabeads m
‑
270streptavidin磁珠,使用200ul 1x bead wash buffer漂洗2次。使用100ul1x bead wash buffer重悬。
78.将杂交后的反应液加入100ul磁珠中,混匀,瞬时离心。pcr仪上65℃杂交45分钟。每15分钟使磁珠重悬一次。
79.加入100ul 65℃预热的1x wash buffer i,在磁力架上去掉上清。
80.加入200ul 65℃预热的1x stringent wash buffer,混匀后65℃5分钟。在磁力架上去掉上清。重复一次。
81.加入200ul室温的1x wash buffer i,混匀。在磁力架上去掉上清。
82.加入200ul室温的1x wash buffer ii,混匀。在磁力架上去掉上清。
83.加入200ul室温的1x wash buffer iii,混匀。在磁力架上去掉上清。
84.加入20ul去离子水重悬磁珠。在磁力架上保留上清,弃掉磁珠。
85.4.目标区域片段3’端连接
86.辅助子a1序列:
87.gtgactggagttcagacgtgtctcttccgatctttttrg(3’端有rna碱基g封闭),序列见seq id no.3,
88.反应体系如下:
[0089][0090]
先将步骤3中得到的捕获产物加去离子水补至30ul。pcr仪上95℃1分钟,4℃保存。
[0091]
将上述20ul反应混合液加入上述dna中,混匀。
[0092]
pcr仪上37℃10分钟,95℃2分钟,72℃5分钟,4℃保存。
[0093]
5.目标区域文库扩增
[0094]
扩增引物f序列:
[0095]
aatgatacggcgaccaccgagatctacactcgtcggcagcgtcagatgtgtataagagacag,序列见seq id no.4,
[0096]
扩增引物r序列:
[0097]
caagcagaagacggcatacgagattccgcgtgtctcgtgggctcgg,序列见seq id no.5,
[0098]
配制如下反应体系:
[0099]
步骤4连接产物50ul5x buffer a20ul扩增引物f(20um)1ul扩增引物r(20um)1uldntp(10mm)1ul2g robust dna聚合酶(5u/ul)1ul去离子水补至100ul
[0100]
扩增程序如下:
[0101][0102]
取5ul进行2%琼脂糖胶电泳。结果见图8。
[0103]
实施例2
[0104]
单样本含标签(umi)目标区域文库构建
[0105]
取人白细胞基因组dna 200ng,使用本发明方法构建含标签的目标区域文库(s2)。
[0106]
1.dna片段化及加标签
[0107]
转座酶识别核心序列:
[0108]
tup3:
[0109]
aatgatacggcgaccaccgagatctacacnnnnnntcgtcggcagcgtcagatgtgtataagagacag,序列见seq id no.6,n表示a、t、g或c碱基。
[0110]
tup2:
[0111]
ctgtctcttatacacatct
[0112]
将tup2和tup3按等摩尔比进行退火,形成双链dna接头tupu。
[0113]
将转座酶和双链dna接头tupu按标准流程组装在一起形成转座酶复合物备用。
[0114]
配制如下反应体系:
[0115]
tupu(50um)4ultn5转座酶(1u/ul)5ul10x tps buffer1ul总体积10ul
[0116]
pcr仪上25℃70min,4℃保存。转座酶复合物即组装完成。
[0117]
dna片段化反应体系如下:
[0118]
dna200ng转座酶复合物2ul5x lm buffer2ul去离子水补至10ul
[0119]
pcr仪上55℃10min,4℃保存。
[0120]
2x ampure xp beads纯化。
[0121]
2.目标区域探针捕获
[0122]
配制反应体系如下:
[0123]
片段化dna20ulcot
‑
1dna5ug
[0124]
将上述反应体系在真空旋转蒸发仪中蒸干液体。
[0125]
加入8.5ul nimblegen 2x杂交缓冲液,3.4ul nimblegen hybridization component a,1.1ul无酶水
[0126]
配制反应体系如下:
[0127][0128][0129]
使用配制好的13ul液体体系重悬蒸干后的dna。转移到0.2ml pcr管中。
[0130]
pcr仪上95℃10分钟。
[0131]
加入4ul xgen pan
‑
cancer panel探针,混匀后瞬时离心。
[0132]
pcr仪上65℃杂交4小时。
[0133]
3.漂洗
[0134]
取100ul dynabeads m
‑
270streptavidin磁珠,使用200ul 1x bead wash buffer漂洗2次。使用100ul1x bead wash buffer重悬。
[0135]
将杂交后的反应液加入100ul磁珠中,混匀,瞬时离心。pcr仪上65℃杂交45分钟。每15分钟使磁珠重悬一次。
[0136]
加入100ul 65℃预热的1x wash buffer i,在磁力架上去掉上清。
[0137]
加入200ul 65℃预热的1x stringent wash buffer,混匀后65℃5分钟。在磁力架上去掉上清。重复一次。
[0138]
加入200ul室温的1x wash buffer i,混匀。在磁力架上去掉上清。
[0139]
加入200ul室温的1x wash buffer ii,混匀。在磁力架上去掉上清。
[0140]
加入200ul室温的1x wash buffer iii,混匀。在磁力架上去掉上清。
[0141]
加入20ul去离子水重悬磁珠。在磁力架上保留上清,弃掉磁珠。
[0142]
4.目标区域片段3’端连接
[0143]
辅助子a1序列:
[0144]
gtgactggagttcagacgtgtctcttccgatctttttrg
[0145]
反应体系如下:
[0146]
[0147][0148]
先将步骤3中得到的捕获产物加去离子水补至30ul。pcr仪上95℃1分钟,4℃保存。
[0149]
将上述20ul反应混合液加入上述dna中,混匀。
[0150]
pcr仪上37℃10分钟,95℃2分钟,72℃5分钟,4℃保存。
[0151]
5.目标区域文库扩增
[0152]
扩增引物f1序列:
[0153]
aatgatacggcgaccaccgagatctacac序列见seq id no.7,扩增引物r序列:
[0154]
caagcagaagacggcatacgagattccgcgtgtctcgtgggctcgg
[0155]
配制如下反应体系:
[0156]
步骤4连接产物50ul5x buffer a20ul扩增引物f1(20um)1ul扩增引物r(20um)1uldntp(10mm)1ul2g robust dna聚合酶(5u/ul)1ul去离子水补至100ul
[0157]
扩增程序如下:
[0158][0159]
取5ul进行2%琼脂糖胶电泳。结果见图9。
[0160]
实施例3
[0161]
多样本含标签(umi)目标区域文库构建
[0162]
取人白细胞基因组dna,每个样本使用30ng,使用本发明方法构建6个含标签的目标区域文库(s1
‑
s6)。
[0163]
1.dna片段化及加标签
[0164]
转座酶识别核心序列:
[0165]
tup3
‑
1:
[0166]
aatgatacggcgaccaccgagatctacacagnnnntcgtcggcagcgtcagatgtgtataagagacag序列见seq id no.8,
[0167]
tup3
‑
2:
[0168]
aatgatacggcgaccaccgagatctacactcnnnntcgtcggcagcgtcagatgtgtataagagacag序列见seq id no.9,
[0169]
tup3
‑
3:
[0170]
aatgatacggcgaccaccgagatctacacgtnnnntcgtcggcagcgtcagatgtgtataagagacag序列见seq id no.10,
[0171]
tup3
‑
4:
[0172]
aatgatacggcgaccaccgagatctacaccannnntcgtcggcagcgtcagatgtgtataagagacag序列见seq id no.11,
[0173]
tup3
‑
5:
[0174]
aatgatacggcgaccaccgagatctacactannnntcgtcggcagcgtcagatgtgtataagagacag序列见seq id no.12,
[0175]
tup3
‑
6:
[0176]
aatgatacggcgaccaccgagatctacacgcnnnntcgtcggcagcgtcagatgtgtataagagacag序列见seq id no.13,
[0177]
上述序列中n均表示a、t、g或c碱基。
[0178]
tup2:
[0179]
ctgtctcttatacacatct
[0180]
将tup3
‑
1、tup3
‑
12、tup3
‑
3、tup3
‑
4、tup3
‑
5、tup3
‑
6分别和tup2按等摩尔比进行退火,分别形成双链dna接头tupu1、tupu2、tupu3、tupu4、tupu5、tupu6。
[0181]
将转座酶分别和双链dna接头tupu1
‑
tupu6按标准流程组装在一起形成6种转座酶复合物备用。
[0182]
配制如下反应体系:
[0183]
tupu(50um)4ultn5转座酶(1u/ul)5ul10x tps buffer1ul总体积10ul
[0184]
pcr仪上25℃70min,4℃保存。转座酶复合物即组装完成。
[0185]
dna片段化反应体系如下:
[0186]
dna50ng转座酶复合物2ul5x lm buffer2ul去离子水补至10ul
[0187]
pcr仪上55℃10min,4℃保存。
[0188]
2x ampure xp beads纯化。
[0189]
2.目标区域探针捕获
[0190]
将6种片段化及加标签产物等体积混合在一起。
[0191]
配制反应体系如下:
[0192]
片段化dna60ulcot
‑
1 dna5ug
[0193]
将上述反应体系在真空旋转蒸发仪中蒸干液体。
[0194]
加入8.5ul nimblegen 2x杂交缓冲液,3.4ul nimblegen hybridization component a,1.1ul无酶水
[0195]
配制反应体系如下:
[0196][0197][0198]
使用配制好的13ul液体体系重悬蒸干后的dna。转移到0.2ml pcr管中。
[0199]
pcr仪上95℃10分钟。
[0200]
加入4ul xgen pan
‑
cancer panel探针,混匀后瞬时离心。
[0201]
pcr仪上65℃杂交4小时。
[0202]
3.漂洗
[0203]
取100ul dynabeads m
‑
270streptavidin磁珠,使用200ul 1x bead wash buffer漂洗2次。使用100ul1x bead wash buffer重悬。
[0204]
将杂交后的反应液加入100ul磁珠中,混匀,瞬时离心。pcr仪上65℃杂交45分钟。每15分钟使磁珠重悬一次。
[0205]
加入100ul 65℃预热的1x wash buffer i,在磁力架上去掉上清。
[0206]
加入200ul 65℃预热的1x stringent wash buffer,混匀后65℃5分钟。在磁力架上去掉上清。重复一次。
[0207]
加入200ul室温的1x wash buffer i,混匀。在磁力架上去掉上清。
[0208]
加入200ul室温的1x wash buffer ii,混匀。在磁力架上去掉上清。
[0209]
加入200ul室温的1x wash buffer iii,混匀。在磁力架上去掉上清。
[0210]
加入20ul去离子水重悬磁珠。在磁力架上保留上清,弃掉磁珠。
[0211]
4.目标区域片段3’端连接
[0212]
辅助子a1序列:
[0213]
gtgactggagttcagacgtgtctcttccgatctttttrg
[0214]
反应体系如下:
[0215][0216][0217]
先将步骤3中得到的捕获产物加去离子水补至30ul。pcr仪上95℃1分钟,4℃保存。
[0218]
将上述20ul反应混合液加入上述dna中,混匀。
[0219]
pcr仪上37℃10分钟,95℃2分钟,72℃5分钟,4℃保存。
[0220]
5.目标区域文库扩增
[0221]
扩增引物f1序列:
[0222]
aatgatacggcgaccaccgagatctacac
[0223]
扩增引物r序列:
[0224]
caagcagaagacggcatacgagattccgcgtgtctcgtgggctcgg
[0225]
配制如下反应体系:
[0226]
步骤4连接产物50ul5x buffer a20ul扩增引物f1(20um)1ul扩增引物r(20um)1uldntp(10mm)1ul2g robust dna聚合酶(5u/ul)1ul去离子水补至100ul
[0227]
扩增程序如下:
[0228][0229][0230]
取5ul进行2%琼脂糖胶电泳。
[0231]
6.上机测序:将文库进行质检后进行illumina高通量测序平台测序。pe150bp测序。
[0232][0233]
实施例4
[0234]
实施例1的基础上探索普通dntp浓度对3’端加尾碱基数量的影响
[0235]
取一段30nt的人工合成单链dna寡核苷酸。
[0236]
依照加入单核苷酸浓度的不同做如下实验。
[0237]
反应体系如下:
[0238][0239]
先将30ul(浓度10um)30nt的人工合成单链dna寡核苷酸在pcr仪上95℃1分钟,4℃保存。
[0240]
将上述20ul反应混合液加入上述dna中,混匀。
[0241]
pcr仪上37℃10分钟,95℃2分钟,72℃5分钟,4℃保存。
[0242]
将产物跑毛细管电泳,通过观察单链dna长度大小可以观察到不同的单核苷酸(datp)浓度对加尾碱基数量的影响。
[0243]
实施例4实验结果见图11。
[0244]
实施例4结果说明加尾碱基的数量与体系中单核苷酸的浓度有关系。浓度越高加尾的数量越多。而加尾数量的过多在ngs测序中会占据大量的数据量。这些数据属于无意义数据。因此,选择合适的单核苷酸的量,对于高效的反应是重要的。
[0245]
实施例5
[0246]
实施例1的基础上探索辅助子的量对3’端加尾碱基数量的影响
[0247]
取一段30nt的人工合成单链dna寡核苷酸。
[0248]
依照加入辅助子的不同比例做如下实验。
[0249]
反应体系如下:
[0250]
[0251]
先将30ul(浓度10um)30nt的人工合成单链dna寡核苷酸在pcr仪上95℃1分钟,4℃保存。
[0252]
将上述20ul反应混合液加入上述dna中,混匀。
[0253]
pcr仪上37℃10分钟,95℃2分钟,72℃5分钟,4℃保存。
[0254]
将产物跑毛细管电泳,通过观察单链dna长度大小可以观察到不同的辅助子和连接子的比例对加尾碱基数量的影响。
[0255]
实施例5实验结果见图12。
[0256]
实施例5结果说明加尾碱基的数量与辅助子在体系中的量有关系。辅助子在一定范围内浓度越高加尾的数量越少。因此,在本发明的实际应用中,选择合适的辅助子浓度对于高效的反应是重要的;这有助于提前终止3’端加尾反应。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1