吡咯并三嗪衍生物及其制备方法和应用与流程
1.本发明属于药物化学技术领域,尤其涉及吡咯并三嗪衍生物及其制备方法和应用。
背景技术:
2.癌症是当今世界严重危害人类健康和生命的疾病之一。随着肿瘤分子生物学研究的不断深入,恶性肿瘤的细胞内信号转导,细胞周期调控和诱导细胞凋亡,血管生成以及细胞外基质和细胞的相互作用等被逐渐阐述清楚。其中,受体酪氨酸激酶(tyrosine kinases receptors,rtks)与肿瘤的发生和发展密切相关。其作用包括激活下游信号转导分子,促进细胞增殖、迁移、存活等。因此,rtks成为抗肿瘤治疗中令人关注的分子治疗靶点。
3.c
‑
met是一类由二硫键链接的异二聚体的受体酪氨酸激酶,它在人体的正常细胞和恶性肿瘤细胞中都存在表达。在遗传性和继发性肾癌、肝癌和其他多种肿瘤中都发现了c
‑
met受体酪氨酸激酶的突变。c
‑
met
‑
hgf/sf信号通路在胚胎发育和组织再生中发挥重要的生理作用。在正常细胞中,c
‑
met
‑
hgf/sf信号通路受到严格的调控;而在肿瘤细胞中,却出现了调节异常。大量研究表明,肿瘤组织中的c
‑
met可以和多种信号分子进行功能性的相互作用,这已成为肿瘤癌变和产生治疗抵抗的重要原因。
4.axl是tam(tyro3、axl、mer)受体酪氨酸激酶(rtk)家族的成员。该激酶家族最初被鉴定为来自于患有慢性粒细胞白血病或慢性骨髓增殖性疾病的患者的细胞中表达的转化基因。axl的激活,通过其同源蛋白配体生长停止特异性蛋白6(gas6)的结合,通过其细胞外结构域的同型二聚化或经白介素(il)
‑
15受体或her2的串扰来进行。axl信号传导刺激细胞应答,包括pi3k
‑
akt、细胞外信号调节的激酶(erk)和p38有丝分裂原激活的蛋白激酶级联的激活,nf
‑
κβ途径,和信号转导子和转录活化子(stat)信号传导。axl信号传导的人量生物学结果,包括侵入、迁移、存活信号传导、血管发生、对化疗和靶向药物的抗性、细胞转化和增殖。此外,axl过表达是导致患者对肿瘤化疗药物或靶向药物产生耐药性的重要原因之一。
5.在癌症治疗过程中,肿瘤转移及耐药性是影响抗癌药物疗效的两大难点,也是导致癌症死亡率居高不下的主要原因。axl表达上调与肿瘤转移的病理机制密切相关。诸多研究结果显示,抑制axl激酶的活性可以有效的阻滞肿瘤细胞的生长、迁移和侵袭。因此,axl激酶抑制剂有可能用于早期癌症患者,尤其是那些容易出现癌细胞转移的患者中,从而最大程度的发挥axl激酶抑制剂的疗效。使用受体酪氨酸激酶抑制剂治疗患者时造成耐药的机制一般为靶向激酶的二次突变或其他受体酪氨酸激酶的代偿性上调。axl激酶的超表达被认为是代偿性上调所产生耐药性的一个重要原因。当一个靶向药物出现耐药性之后将其同axl激酶抑制剂联合使用会产生药效的协同效果,并且这种效果已经在细胞水平上和动物体内的多个肿瘤模型中得到了验证。除此之外,肿瘤细胞也可以通过上皮细胞
‑
间充质转化机制产生耐药性。晚期癌症患者通常需要二线甚至三线用药来减少其对一线的抗肿瘤药物的耐药性,在这种情况下,axl抑制剂可以通过和一线药物联用的方式来减缓患者的耐药
性,从而达到抑制癌症进展的效果。因此,开发一种axl和c
‑
met双靶点抑制剂存在迫切需求。
技术实现要素:
6.本发明旨在至少解决上述现有技术中存在的技术问题之一。为此,本发明第一个方面提出吡咯并三嗪衍生物,能够同时对c
‑
met和axl靶点有较好的抑制作用。
7.本发明的第二个方面提出了包含上述吡咯并三嗪衍生物的药物组合物。
8.本发明的第三个方面提出了上述吡咯并三嗪衍生物的制备方法。
9.本发明的第四个方面提出了上述吡咯并三嗪衍生物的应用。
10.根据本发明的第一个方面,提出了通式i化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,
[0011][0012]
其中:r1选自h或卤素;
[0013]
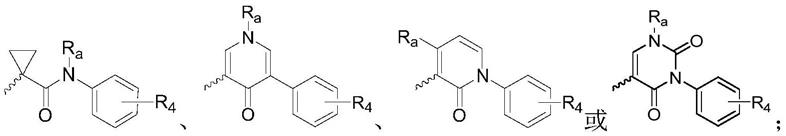
r2选自
[0014]
r3选自任意地被一个或多个r
b
取代的杂芳基;所述杂芳基选自吡唑基、吡啶基、咪唑基、噻吩基、呋喃基或噻唑基;
[0015]
r4选自h、卤素或烷氧基;
[0016]
r
a
选自h、c1~c6烷基或c1~c6烷氧基;
[0017]
r
b
选自c1~c4烷基、c3~c6环烷基、c3~c6杂环烷基或
‑
so2r
c
;所述的c1~c4烷基、c3~c6环烷基、c3~c6杂环烷基可被一个或多个羟基、氨基、卤素或烷氧基取代,r
c
可为c1~c4烷基、c3~c6环烷基或苯基;
[0018]
n选自1~5的任意整数。
[0019]
在本发明的一些实施方式中,r3选自
[0020]
在本发明的一些优选的实施方式中,r
b
选自
[0021]
在本发明的一些更优选的实施方式中,所述通式i化合物选自以下化合物:
[0022]
[0023][0024]
根据本发明的第二个方面,提出了一种药物组合物,包括所述通式i化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药。
[0025]
在本发明的一些实施方式中,所述药物组合物还包括选自下列组中的一种或多种:酪氨酸蛋白酶抑制剂、egfr抑制剂、vegfr抑制剂、bcr
‑
abl抑制剂、c
‑
kit抑制剂、c
‑
met抑制剂、raf抑制剂、mek抑制剂、组蛋白去乙酰酶抑制剂、vegf抗体、egf抗体、hiv蛋白激酶抑制剂、hmg
‑
coa还原酶抑制剂、pd
‑
1抑制剂、pd
‑
l1抑制剂等。
[0026]
在本发明的一些优选的实施方式中,可以将本发明的化合物、异构体、溶剂合物、结晶或前药与药学上可接受的载体、稀释剂或赋形剂混合制备成药物制剂,以适合于经口或胃肠外给药。给药方法包括但不限于皮内、肌内、腹膜内、静脉内、皮下、鼻内和经口途径。所述制剂可以通过任何途径施用,例如通过输注或推注,通过经上皮或皮肤粘膜(例如口腔粘膜或直肠等)吸收的途径施用。给药可以是全身的或局部的。经口施用制剂的实例包括固体或液体剂型,具体而言,包括片剂、丸剂、粒剂、粉剂、胶囊剂、糖浆、乳剂、混悬剂等。所述制剂可通过本领域已知的方法制备,且包含药物制剂领域常规使用的载体、稀释剂或赋形
剂。
[0027]
根据本发明的第三个方面,提出了本发明的通式i化合物的制备方法:包括以下步骤:
[0028]
式ii化合物与式iii化合物发生取代反应后,再与式iv化合物进行偶联反应,制得通式i化合物;其中,r1、r2、r3的定义如上述通式i化合物中的定义。
[0029]
在本发明的一些实施方式中,所述式ii化合物的制备方法为:式v化合物与式vi化合物发生缩合反应制备得到。
[0030]
在本发明的一些优选的实施方式中,所述式ii化合物与式iii化合物发生取代反应制得,产物是式vii化合物
[0031]
在本发明的一些更优选的实施方式中,所述偶联反应的催化剂选自pd(dppf)cl2、pd(oac)2、pd2(dba)3、pd(pph3)2cl2、pd(pph3)4、xphos
‑
pd
‑
g3、xphos
‑
pd
‑
g2、xphos
‑
pd
‑
g1、ruphos
‑
pd
‑
g3、sphos
‑
pd
‑
g2等市售钯催化剂的至少一种。
[0032]
在本发明的一些更优选的实施方式中,所述偶联反应的碱选自碳酸钠、碳酸钾、碳酸铯、磷酸钾、醋酸钾、dbu、三乙胺、n,n
‑‑
二异丙基乙胺、吡啶中的至少一种;优选为碳酸铯。
[0033]
根据本发明的第四个方面,提出了本发明的通式i化合物、异构体、溶剂合物、结晶或前药或本发明的药物组合物在制备治疗或预防肿瘤药物中的应用。
[0034]
在本发明的一些实施方式中,所述肿瘤包括肺癌、胃癌、肝癌、肾癌、食管癌、乳腺
癌、白血病、前列腺癌、结直肠癌、骨癌、大肠癌、黑色素瘤、淋巴瘤、血癌、脑瘤、胰腺癌或皮肤癌中的至少一种。
[0035]
本发明的“溶剂合物”在常规意义上是指溶质(如活性化合物、活性化合物的盐)和溶剂(如水)组合形成的复合物。溶剂是指本领域的技术人员所知的或容易确定的溶剂。如果是水,则溶剂合物通常被称作水合物,例如一水合物、二水合物、三水合物等。
[0036]
本发明的“结晶”是指本发明所述的化合物形成的各种固体形态,包括晶型、无定形。
[0037]
本发明的“异构体”包括化合物构型异构体、构象异构体和对映异构体。构型异体是指顺式或反式构型的顺反异构体;构象异构体是指由于单键旋转产生的立体异构体。
[0038]
本发明的“前药”是指在生物体的生理条件下,由于与酶、胃酸等反应而转化成本发明的化合物,即通过酶的氧化、还原、水解等转化成本发明的化合物和/或通过胃酸等的水解反应等转化成本发明的化合物。
[0039]
本发明的“药学可接受的盐”是指本发明的化合物与酸形成的药学上可接受的盐,所述的酸包括但不限于磷酸、硫酸、盐酸、氢溴酸、柠檬酸、马来酸、丙二酸、扁桃酸、琥珀酸、富马酸、醋酸、乳酸、硝酸等等。
[0040]
本发明的“药物组合物”是指包含任何一种本文所述的化合物,包括异构体、前药、溶剂合物、药学上可接受的盐或其化学的保护形式,和一种或多种药学上可接受载体的混合物。
[0041]
本发明的“药学上可接受的载体”是指对有机体不引起明显刺激性和不干扰所给予化合物的生物活性和性质的载体,包含溶剂、稀释剂或其它赋形剂、分散剂、表面活性剂、等渗剂、增稠剂或乳化剂、防腐剂、固体粘合剂、润滑剂等。除非任何常规载体介质与本发明化合物不相容。可以作为药学上可接受的载体的一些实例包括,但不限于糖类,如乳糖、葡萄糖和蔗糖;淀粉,如玉米淀粉和马铃薯淀粉;纤维素及其衍生物,如羧甲基纤维素钠、以及纤维素和乙酸纤维素;麦芽、明胶等。
[0042]
本发明的“赋形剂”指加入到药用组合物中以进一步促进给予化合物的惰性物质。赋形剂可以包括碳酸钙、磷酸钙、多种糖类和多种类型的淀粉、纤维素衍生物、明胶、植物油、聚乙二醇。
[0043]
本发明的“在制备用于治疗或预防肿瘤的药物中的应用”是指可以抑制肿瘤的生长、发展和/或转移,主要向所需要的人或动物给治予治疗有效剂量的本发明的化合物以抑制、减慢或逆转受治疗者肿瘤的生长、迁移或扩散。
[0044]
本发明的有益效果为:
[0045]
1.本发明化合物能够有效地抑制axl和c
‑
met激酶的活性。
[0046]
2.本发明化合物能够有效抑制肿瘤细胞的体内生长,并具有良好的安全性和耐受性。
[0047]
3.本发明化合物的制备方法工艺简单,便于工业化生产。
附图说明
[0048]
下面结合附图和实施例对本发明做进一步的说明,其中:
[0049]
图1为本发明试验例2中化合物组、溶剂对照组和阳性对照组小鼠肿瘤体积的生长
变化图。
[0050]
图2为本发明试验例2中化合物组、溶剂对照组和阳性对照组小鼠体重随治疗时间的变化图。
具体实施方式
[0051]
以下将结合实施例对本发明的构思及产生的技术效果进行清楚、完整地描述,以充分地理解本发明的目的、特征和效果。显然,所描述的实施例只是本发明的一部分实施例,而不是全部实施例,基于本发明的实施例,本领域的技术人员在不付出创造性劳动的前提下所获得的其他实施例,均属于本发明保护的范围。
[0052]
实施例1
[0053]
本实施例制备了一种n
‑
(4
‑
(6
‑
(1
‑
甲基
‑
1h
‑
吡唑
‑4‑
基)吡咯并[2,1
‑
f][1,2,4]三嗪
‑4‑
基)氧基)苯基)
‑
n
‑
(4
‑
氟苯基)环丙烷
‑
1,1
‑
二甲基酰胺,具体过程为:
[0054][0055]6‑
溴
‑4‑
氯吡咯并[2,1
‑
f][1,2,4]三嗪(920mg,3.96mmol,1.0eq)、n
‑
(4
‑
氟苯基)
‑
n
‑
(4
‑
羟基苯基)环丙烷
‑
1,1
‑
二甲基酰胺(1.31g,4.16mmol,1.05eq)和碳酸钾(820mg,5.94mmol,1.5eq)于常温加入到反应瓶中,用dmf(12ml)溶成悬浮液,换氮气3次,移入80℃油浴锅中搅拌6小时。
[0056]
反应液冷却至20℃,缓慢倒入到饱和氯化铵溶液(60ml)中,加乙酸乙酯萃取(20ml*5),饱和食盐水(40ml)洗涤有机相,硫酸钠干燥后,40℃下减压浓缩,再进行柱层析纯化,制得1.30g白色固体。
[0057]
lcms(esi):m/z 509/511[m+h]
+
。
[0058][0059]
n
‑
(4
‑
((6
‑
溴吡咯并[2,1
‑
f][1,2,4]三嗪
‑4‑
基)氧)苯基)
‑
n
‑
(4
‑
氟苯基)环丙烷
‑
1,1
‑
二甲基酰胺(500mg,0.980mmol,1.0eq),1
‑
甲基
‑4‑
吡唑硼酸嚬哪醇酯(214mg,1.03mmol,1.05eq),碳酸铯(957mg,2.94mmol,3.0eq),pd(dppf)cl2(41.5mg)常温加入到反应瓶中,用异丁醇(4ml)和水(2ml)溶成悬浮液,换氮气3次,移入85℃油浴锅中搅拌13小时。tlc监测(展开剂比例为石油醚:乙酸乙酯=1:1,r
f
(产物)=0.25)。
[0060]
反应液冷却至20℃,缓慢倒入到饱和edta
‑
edta二钠盐缓冲液(20ml)中,加乙酸乙酯萃取(15ml*4),饱和食盐水(30ml)洗涤有机相,硫酸钠干燥后,40℃下减压浓缩,得粗产品。粗产品加入到二甲基甲酰胺(4ml)和乙腈(4ml)混合液中溶解,过液相色谱柱,反相分离单针1ml;流动相为4
‰
氨水:乙腈,梯度洗脱:0~30min,50~100%,目标产物保留时间
18min,80%乙腈。柱型号(大曹c18
‑
100
‑
8um,id 25.4mm*450mm)。馏分于40℃减压浓缩,冷冻干燥17.5小时。制得23mg白色固体。
[0061]
lcms(esi):m/z 511[m+h]
+
。1h nmr(500mhz,dmso
‑
d6)δ10.12(s,1h),10.02(s,1h),8.30(d,j=1.6hz,1h),8.12(s,1h),8.05(s,1h),7.90(s,1h),7.73
–
7.68(m,2h),7.62(dd,j=8.9,5.1hz,2h),7.30
–
7.20(m,3h),7.14(t,j=8.9hz,2h),3.90(q,j=5.5hz,3h),1.46(s,4h).
[0062]
实施例2
[0063]
本实施例制备了一种n
‑
(4
‑
(6
‑
(1
‑
(2
‑
羟乙基)
‑
1h
‑
吡唑
‑4‑
基)吡咯并[2,1
‑
f][1,2,4]三嗪
‑4‑
基)氧基)苯基)
‑
n
‑
(4
‑
氟苯基)环丙烷
‑
1,1
‑
二甲基酰胺,具体过程为:
[0064][0065]
n
‑
(4
‑
((6
‑
溴吡咯并[2,1
‑
f][1,2,4]三嗪
‑4‑
基)氧)苯基)
‑
n
‑
(4
‑
氟苯基)环丙烷
‑
1,1
‑
二甲基酰胺(600mg,1.18mmol,1.0eq)、1
‑
(羟乙基)吡唑
‑4‑
硼酸频哪醇酯(294mg,1.24mmol,1.05eq)、碳酸铯(1.15g,3.54mmol,3.0eq)和pd(dppf)cl2(100mg)于常温加入到反应瓶中,用叔戊醇(6ml)和水(1.8ml)溶成悬浮液,换氮气3次,移入97℃油浴锅中搅拌6.5小时。
[0066]
反应液冷却至20℃,缓慢倒入到饱和氯化铵溶液(50ml)中,加乙酸乙酯萃取(15ml*7),饱和食盐水(40ml)洗涤有机相,硫酸钠干燥后,40℃下减压浓缩,再进行柱层析纯化,制得284mg类白色固体。
[0067]
lcms(esi):m/z 542[m+h]
+
。1h nmr(500mhz,dmso
‑
d6)δ10.14(s,1h),10.04(s,1h),8.32(d,j=1.6hz,1h),8.14(s,1h),8.05(s,1h),7.91(s,1h),7.73
–
7.68(m,2h),7.63(dd,j=8.9,5.1hz,2h),7.30
–
7.22(m,3h),7.14(t,j=8.9hz,2h),4.92(t,j=5.2hz,1h),4.16(t,j=5.6hz,2h),3.77(q,j=5.5hz,2h),1.48(s,4h).
[0068]
试验例1
[0069]
本试验例测试化合物对激酶axl和c
‑
met的酶活性抑制性(ic
50
)
[0070]
本实验使用迁移率变动分析法(mobility shift assay)的方法,在axl和c
‑
met激酶上进行化合物的筛选,起始浓度10000nm,3倍稀释,10个浓度,复孔检测。
[0071]
试剂及耗材如表1:
[0072]
表1
[0073]
[0074][0075]
仪器:
[0076]
离心机(生产厂家:eppendorf,型号:5430)
[0077]
酶标仪(生产厂家:perkin elmer,型号:caliper ez readerⅱ)
[0078]
echo 550(生产厂家:labcyte,型号:echo 550)
[0079]
酶标仪(生产厂家:perkin elmer,型号:envision)
[0080]
具体过程为:
[0081]
1)配制1
×
激酶缓冲液;
[0082]
2)化合物浓度梯度的配制:受试化合物测试浓度为10000nm,3倍稀释,10个浓度,复孔检测;在384目的板中稀释成100倍终浓度的100%dmso溶液,3倍稀释化合物,10个浓度。使用分液器echo 550向目的板384孔板转移250nl的100倍终浓度的化合物。
[0083]
3)用1
×
激酶缓冲液配制2.5倍终浓度的激酶溶液。
[0084]
4)在化合物孔和阳性对照孔分别加10μl的2.5倍终浓度的激酶溶液;在阴性对照孔中加10μl的1
×
激酶缓冲液。
[0085]
5)1000rpm离心30秒,反应板振荡混匀后室温孵育10分钟。
[0086]
6)用1
×
激酶缓冲液配制5/3倍终浓度的atp和激酶底物的混合溶液。
[0087]
7)加入15μl的5/3倍终浓度的atp和底物的混合溶液,起始反应。
[0088]
8)将384孔板1000rpm离心30秒,振荡混匀后室温孵育相应的时间。
[0089]
9)加入30μl终止检测液停止激酶反应,1000rpm离心30秒,振荡混匀。
[0090]
10)用酶标仪(caliper ez reader)读取转化率。
[0091]
采用分析软件graphpad prism 5的log(inhibitor)vs.response
‑
variable slope拟合量效曲线,从而得出各个化合物对酶活性的ic
50
值。结果如表2所示。
[0092]
表2
[0093][0094]
由上表可知,本发明化合物能够有效地抑制axl和c
‑
met激酶的活性。与阳性对照卡博替尼、司曲替尼相比,实施例2化合物展现出略高或相当的活性抑制能力。
[0095]
试验例2
[0096]
本试验例测试化合物在模型小鼠肿瘤细胞异位移植试验中的体内抗肿瘤活性情况,具体过程为:
[0097]
选择12只雌性6
‑
8周龄的balb/c裸小鼠,皮下异位接种人肺癌细胞系a549肿瘤细胞株5
×
106个,接种肿瘤细胞26天后,待肿瘤长至60mm3~250mm3时,小鼠灌胃给予待测试样品。
[0098]
将小鼠分为阴性溶剂对照组、本发明化合物组(实施例2化合物)(20mg/kg)和阳性对照司曲替尼组(20mg/kg),每组4只。所有剂量组均采用等体积不等浓度经口每日单次灌胃给药,给药体积为10ml/kg。阴性溶剂对照组给予相同体积的空白溶媒(dmso:solutol:水=1:2:7),给药频率为每天一次,连续给药14天。
[0099]
开始给药后,每周测量两次小鼠的体重和肿瘤的大小。肿瘤大小计算公式:
[0100]
肿瘤体积(mm3)=0.5
×
(肿瘤长径
×
肿瘤短径2)。
[0101]
抗肿瘤疗效是基于治疗中肿瘤的生长曲线(即每次测量的肿瘤体积相对于其治疗天数)和相对瘤体积来评估。其中相对肿瘤抑制率(tgi)按下列公式计算:
[0102]
相对肿瘤抑制率tgi(%):tgi%=(1
‑
t/c)
×
100%。
[0103]
t/c%为相对肿瘤增值率,即在某一时间点,治疗组和对照组相对肿瘤体积或瘤重的百分比值。t和c分别为治疗组和对照组在某一特定时间点的相对肿瘤体积(rtv)或瘤重(tw)。计算公式如下:t/c%=t
rtv
/c
rtv
×
100%(t
rtv
:治疗组平均rtv;c
rtv
:溶媒对照组平均rtv;rtv=vt
‑
v0,v0为分组时该动物的瘤体积,vt为治疗后该动物的瘤体积)。或t/c%=ttw/ctw
×
100%(ttw:治疗组试验终结时平均瘤重;ctw:溶媒对照组试验终结时平均瘤重)。结果如图1和图2所示。
[0104]
图1显示本发明化合物组、溶剂对照组和阳性对照组小鼠肿瘤体积的生长变化。如图所示,本发明化合物能够有效抑制肿瘤细胞在模型小鼠体内的生长,相对肿瘤抑制率(tgi)以体积计为89.02%;以瘤重计为63.95%,高于阳性对照司曲替尼的57.37%。
[0105]
图2显示本发明化合物组、溶剂对照组和阳性对照组小鼠体重随治疗时间的变化。如图所示,荷瘤小鼠在实验过程中体重无明显变化,显示本发明化合物具有良好的安全性和耐受性。
[0106]
上面对本发明实施例作了详细说明,但是本发明不限于上述实施例,在所属技术领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下作出各种变
化。此外,在不冲突的情况下,本发明的实施例及实施例中的特征可以相互组合。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1