一种光刻胶树脂单体及其中间体的制备方法与流程
1.本发明涉及有机合成技术领域,特别是涉及一种光刻胶树脂单体及其中间体的制备方法。
背景技术:
2.光刻胶(photoresist)又称光致抗蚀剂,是指通过紫外光、电子束、离子束、x射线等的照射或辐射,其溶解度发生变化的耐蚀剂刻薄膜材料。由感光树脂、增感剂和溶剂3种主要成分组成的对光敏感的混合液体。在光刻工艺过程中,用作抗腐蚀涂层材料。半导体材料在表面加工时,若采用适当的有选择性的光刻胶,可在表面上得到所需的图像。光刻胶按其形成的图像分类有正性、负性两大类。在光刻胶工艺过程中,涂层曝光、显影后,曝光部分被溶解,未曝光部分留下来,该涂层材料为正性光刻胶。如果曝光部分被保留下来,而未曝光被溶解,该涂层材料为负性光刻胶。按曝光光源和辐射源的不同,又分为紫外光刻胶(包括紫外正、负性光刻胶)、深紫外光刻胶、x-射线胶、电子束胶、离子束胶等。含内酯化合物的光刻胶树脂单体可给予树脂充分且平均的耐蚀刻以及基板密著性,广泛应用于光刻胶的树脂单体,但是现有的制备方法收率低、纯度低。
技术实现要素:
3.鉴于以上所述现有技术的缺点,本发明的目的在于提供一种光刻胶树脂单体及其中间体的制备方法,收率高、纯度高。
4.为实现上述目的及其他相关目的,本发明第一方面提供一种光刻胶树脂中间体的制备方法,包括如下步骤:
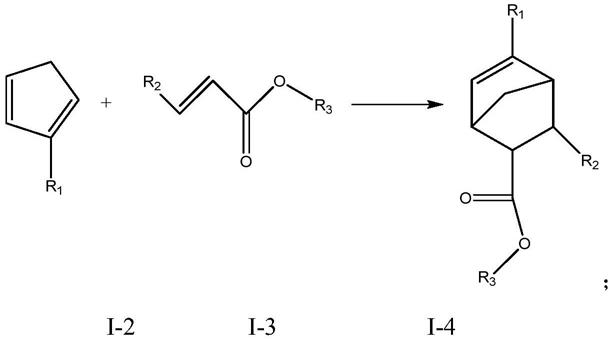
5.a)式i-1化合物进行解聚,获得式i-2化合物,反应路线如下:
[0006][0007]
b)将式i-2化合物与式i-3化合物反应,获得式i-4化合物,反应路线如下:
[0008][0009]
c)将步骤b)的反应产物进行碱化处理、酸化处理和浓缩;
[0010]
其中,r1为氢原子、直链状烷基、支链状烷基或环状烷基中的一种;r2为氢原子或
co2r3;r3为氢原子、直链状烷基、支链状烷基或环状烷基中的一种。
[0011]
r1为碳数1~8的直链状烷基、支链状烷基或环状烷基,如甲基、乙基、丙基、异丙基、正丁基、叔丁基、仲丁基、异丁基、正戊基、叔戊基、正己基、环戊基、环己基、环戊甲基、环戊乙基、环己甲基、环己乙基等。r3为碳数1~15的直链状烷基、支链状烷基或环状烷基,如甲基、乙基、丙基、异丙基、正丁基、叔丁基、仲丁基、异丁基、正戊基、叔戊基、正己基、环戊基、环己基、乙基环戊基、丁基环戊基、乙基环己基、丁基环己基、金刚烷基、乙基金刚烷基、丁基金刚烷基等。
[0012]
优选地,还包括如下技术特征中的至少一项:
[0013]
a1)步骤a)中,解聚的温度为150~200℃,如150~165℃或165~200℃;
[0014]
a2)步骤a)中,解聚的压力为常压;常压是指一个大气压;
[0015]
a3)步骤a)中,解聚收集的馏分为36~42℃馏分;
[0016]
b1)步骤b)中,式i-2化合物与式i-3化合物的摩尔比为1~1.5:1,如1~1.09:1、1.09~1.2:1或1.2~1.5:1;
[0017]
b2)步骤b)中,反应温度为38~42℃;
[0018]
b2)步骤b)在惰性气体保护下进行,如氮气等;
[0019]
c1)步骤c)具体包括如下步骤:
[0020]
c11)将步骤b)的反应产物、第一有机溶剂、碱和水混合,分层,得到水层相;
[0021]
c12)将所述水层相与酸混合后加入第二有机溶剂,分层,得到有机层相;
[0022]
c13)将所述有机层相进行浓缩回收所述第一有机溶剂和所述第二有机溶剂。
[0023]
更优选地,特征c1)中,还包括如下技术特征中的至少一项:
[0024]
c111)步骤c11)中,所述步骤b)的反应产物与所述第一有机溶剂的质量比为2~5:1,如2~3.3:1、3.3~4:1或4~5:1;
[0025]
c112)步骤c11)中,混合后的ph值为12~13;
[0026]
c113)步骤c11)中,所述第一有机溶剂选自甲基叔丁基醚、二氯甲烷或三氯甲烷中的至少一种;
[0027]
c114)步骤c11)中,所述碱选自氢氧化钠或氢氧化钾中的至少一种;
[0028]
c1)步骤c)中,在进行碱化处理前,将步骤b)的反应产物降温至20~30℃;
[0029]
c121)步骤c12)中,所述水层相与所述第二有机溶剂的质量比为1~3:1,如1~2:1或2~3:1;
[0030]
c122)步骤c12)中,混合后的ph值为2~3;
[0031]
c123)步骤c12)中,所述酸选自盐酸、醋酸或磷酸中的至少一种;
[0032]
c131)步骤c13)中,所述有机层控温为35~45℃;
[0033]
c132)步骤c13)中,浓缩的压力为20~50mmhg,如20~30mmhg、30~40mmhg或40~50mmhg。
[0034]
本发明第二方面提供一种光刻胶树脂单体的制备方法,包括如下步骤:
[0035]
1)采用上述光刻胶树脂中间体的制备方法获得式i-4化合物;
[0036]
2)将式i-4化合物与氧化剂进行环氧化反应,获得式i-5化合物,反应路线如下:
[0037][0038]
3)将式i-5化合物进行内酯化反应,获得式i-6化合物,反应路线如下:
[0039][0040]
4)将式i-6化合物与式i-7化合物进行酯化反应,获得式i化合物,反应路线如下:
[0041][0042]
其中,r4为氢原子、甲基或co2r3;r5为氢原子、甲基或ch2co2r3。
[0043]
优选地,步骤2)中,还包括如下技术特征中的至少一项:
[0044]
21)式i-4化合物与所述氧化剂的摩尔比为1:1.0~2.0,如1:1.0~1.35或1:1.35~2.0;
[0045]
22)环氧化反应的温度为57~63℃;
[0046]
23)所述氧化剂选自双氧水、间氯过氧苯甲酸或过氧乙酸中的至少一种;
[0047]
24)在甲酸存在的条件下进行环氧化反应;
[0048]
25)步骤2)还包括如下步骤:将步骤2)的反应产物进行淬灭反应、萃取和浓缩。
[0049]
更优选地,还包括如下技术特征中的至少一项:
[0050]
241)特征24)中,式i-4化合物与甲酸的摩尔比为1:1~2,如1:1~1:1.5或1:1.5~1:2;
[0051]
251)特征25)中,所述淬灭反应采用的淬灭剂选自亚硫酸钠、亚硫酸氢钠或连二亚硫酸钠即连二亚硫酸钠中的至少一种;
[0052]
252)特征25)中,所述萃取采用的萃取剂选自乙酸乙酯、二氯甲烷、二氯乙烷、三氯乙烷、甲苯、四氢呋喃或甲基四氢呋喃中的至少一种;
[0053]
253)特征25)中,浓缩时,所述萃取得到的有机层控温在37~43℃;
[0054]
254)特征25)中,浓缩的压力为20~50mmhg,如20~35mmhg、35~45mmhg或45~50mmhg。
[0055]
优选地,步骤3)中,还包括如下技术特征中的至少一项:
[0056]
31)所述内酯化反应在甲醇和碳酸钾存在的条件下进行;
[0057]
32)内酯化反应的温度为15~25℃;
[0058]
33)步骤3)还包括如下步骤:将步骤3)的反应产物进行第一次浓缩、萃取、干燥和第二次浓缩。
[0059]
更优选地,还包括如下技术特征中的至少一项:
[0060]
311)特征31)中,式i-5化合物与甲醇的摩尔比为1:2~5,如1:2~1:3或1:3~1:5;
[0061]
312)特征31)中,式i-5化合物与碳酸钾的摩尔比为1:0.2~1,如1:0.2~1:0.6或1:0.6~1:1;
[0062]
331)特征33)中,第一次浓缩控温为35~45℃;
[0063]
332)特征33)中,第一次浓缩的压力为20~50mmhg,如20~30mmhg、30~40mmhg或40~50mmhg;
[0064]
333)特征33)中,第一次浓缩脱除甲醇;
[0065]
334)特征33)中,加入乙酸乙酯和水进行萃取;
[0066]
335)特征33)中,第二次浓缩控温为38~42℃;
[0067]
336)特征33)中,第二次浓缩的压力为20~50mmhg,如20~30mmhg、30~40mmhg或40~50mmhg;
[0068]
337)特征33)中,第二次浓缩脱除乙酸乙酯。
[0069]
优选地,步骤4)中,还包括如下技术特征中的至少一项:
[0070]
41)式i-6化合物与式i-7化合物的摩尔比为1:1~1.5,如1:1~1:1.2、1:1.2~1:1.3或1:1.3~1:1.5;
[0071]
42)酯化反应的温度为3~7℃;
[0072]
43)所述酯化反应在三乙胺和二氯甲烷存在的条件下进行;
[0073]
44)步骤4)在惰性气体保护下进行;
[0074]
45)进行酯化反应时加入阻聚剂;
[0075]
46)步骤3)还包括如下步骤:将步骤3)的反应产物进行萃取、浓缩、打浆。
[0076]
更优选地,还包括如下技术特征中的至少一项:
[0077]
431)特征43)中,式i-6化合物与三乙胺的摩尔比为1:1.5~2,如1:1.5~1:1.8或1:1.8~1:2;
[0078]
432)特征43)中,式i-6化合物与二氯甲烷的摩尔比为1:5~10,如1:5~1:7或1:7~1:10;
[0079]
451)特征45)中,所述阻聚剂选自吩噻嗪、对甲氧基苯酚、对苯二酚、对苯醌或对叔丁基邻苯二酚中的至少一种;
[0080]
452)特征45)中,式i-6化合物与所述阻聚剂的摩尔比为1:0.01~0.05,如1:0.01~1:0.03或1:0.03~1:0.05;
[0081]
461)特征46)中,将步骤3)的反应产物加入水分层萃取,水相用二氯乙烷提取,合并有机相;
[0082]
462)特征46)中,浓缩脱除二氯甲烷;
[0083]
463)特征46)中,加入甲醇打浆。
[0084]
本发明与现有技术相比,光刻胶树脂中间体的制备方法包括将步骤b)的反应产物进行碱化处理和酸化处理,能够将式i-1化合物除掉,如环戊二烯二聚体(臭味,导致聚合的元凶),光刻胶树脂单体的纯度能提高到99%以上。光刻胶树脂单体及其中间体的制备方法收率高、纯度高,尤其在百公斤级制备下仍然具有收率高和纯度高的有益效果。
附图说明
[0085]
图1是实施例1制备得到的产品核磁图谱。
[0086]
图2是实施例1制备得到的产品的质谱。
具体实施方式
[0087]
下面通过具体实施例,对本发明的技术方案作进一步的具体说明。应当理解,本发明的实施并不局限于下面的实施例,对本发明所做的任何形式上的变通和/或改变都将落入本发明保护范围。
[0088]
在本发明中,所采用的设备和原料等均可从市场购得或是本领域常用的。下述实施例中的方法,如无特别说明,均为本领域的常规方法。
[0089]
实施例1
[0090][0091]
将400kg二聚环戊二烯倒入反应釜中,常压内温165℃左右,馏分36~42℃,接收装置为冰浴冷却,收集到包含1,3-环戊二烯的第一料液。
[0092]
向500l釜内抽入丙烯酸200kg,升温至釜内温度40
±
2℃。釜内氮气保护下,开始滴加第一料液。约14小时滴加完毕。第一料液滴加完毕后,釜内控温40
±
2℃,保温反应6小时,保温结束后取样测gc,要求产品纯度大于90%。反应结束后,循环水降温至25
±
5℃,得到第二料液。碱化处理:将第二料液转入1000l釜内,向釜内抽入甲基叔丁基醚180kg,搅拌10分钟。提前向500l釜内加入水400kg,循环水降温下,缓慢投入氢氧化钠140kg,搅拌溶清,抽入1000l高位槽内。料液釜内控温25
±
5℃,滴加氢氧化钠水溶液,约滴加300l后,釜内搅拌10分钟,取样测ph值,要求ph为12-13合格,ph值调好后,搅拌10分钟,复测ph。ph为12-13时,开始搅拌30分钟,静止1小时,分层(产品位于水层内)。有机层装桶,水层送样测gc,要求杂质峰小于1%为合格。检测合格后,水层入1000l釜内,有机层入500l釜内,常压回收甲基叔丁基醚。酸化处理:向1000l釜高位槽内抽入盐酸300kg,釜内料液控温25
±
2℃,滴加高位槽内盐酸,调ph为2-3,滴加约250kg后,开始取样测ph,若不合格每滴加20kg盐酸后检测一次,至合格。釜内料液ph值调好后,向釜内抽入甲基叔丁基醚400kg(产品位于有机层内),搅拌30分钟,静止30分钟,分层,下层水层入另一个1000l釜内,有机层保留在釜内。有机层控温40
±
5℃,减压浓缩甲基叔丁基醚(回收甲基叔丁基醚可以套用)浓缩至无液体流出后,再继续浓缩3小时,取样测gc,要求溶剂残留小于2%,其中浓缩的压力为50mmhg。浓缩到最后料液
为油状物,产出油状物369kg。
[0093]
向1000l釜内抽入甲酸122kg,抽入油状物369kg,向1000l釜高位槽抽入双氧水369kg。滴加双氧水369kg,约8小时滴加完毕。滴加结束后,控温60
±
3℃,保温反应1小时,保温结束后取样测gc,要求原料小于0.5%。提前在1000l釜内抽入水350kg,搅拌下投入亚硫酸钠110kg,搅拌30分钟后,釜内降温5
±
3℃。釜内控温在5
±
5℃将反应合格的料液转入釜内,约8小时转完。釜内控温15
±
3℃,滴加盐酸调ph值为2-3,ph值调好后,料液称重。使用乙酸乙酯进行萃取多次,分层,有机层合并,水层确定无料后放掉。有机层控温40
±
3℃,减压浓缩至无液体流出,再继续浓缩2小时,浓缩压力为35mmhg,取样测gc,要求溶剂残留小于2%。浓缩结束后降温至常温,釜内料液装入干净桶中称重。产出油状物:435kg。
[0094]
向1000l釜内抽入甲醇480kg,抽入油状物200kg,投入碳酸钾60kg,控温20
±
5℃,搅拌3小时,取样测gc,要求中间态小于5%(中间态位置为主峰后面第一个峰),若不合格每1小时,取样一次至合格为至(反应3小时,中间态为6.02%,反应4小时中间态为4.69%)。反应合格为,釜内控温40
±
5℃,减压浓缩至无液体流出,浓缩压力为40mmhg。釜内浓缩结束后,使用乙酸乙酯进行萃取多次,分层,有机层合并,水层确定无料后放掉。向有机层釜内加入无水硫酸钠25kg,室温搅拌2小时,取样测水份,要求水份小于0.5%。合格为过滤,滤饼用ea20kg淋洗后,过滤,过完后。有机层控温40
±
2℃,减压浓缩至无液体流出,浓缩压力为40mmhg,浓缩结束后釜内有大量白色固体析出,收率为59.25%。
[0095]
向1000l釜内抽入料液610kg、抽入二氯甲烷270kg、抽入三乙胺132kg入釜,抽料结束后,氮气置换,氮气保护下投入对叔丁基邻苯二酚1100g。氮气保护下,釜内控温5
±
2℃开始,滴加甲基丙烯酰氯97kg,约8小时滴加完毕。滴加完后5
±
2℃,保温反应2小时,取样测gc,要求原料小于0.5%。提前向1000l釜内加入水400kg,降温至2
±
2℃,釜内控温在15℃以下,将反应液转入水解釜内,约3小时转完。转完料后,室温搅拌30分钟,静止30分钟,分层,下层有机层分入周转桶内,水层加入二氯甲烷200kg。室温下搅拌30分钟,静置30分钟,分层,水层确定无料后,放掉,有机层合并,加入水300kg,室温搅拌30分钟,静置30分钟,分层,水层确定无料后,放掉。有机层加入无水硫酸钠50kg,室温搅拌2小时,取样测水份,要求水份小于0.5%过滤,过滤滤饼用二氯甲烷30kg,淋洗,过滤。减压除去二氯甲烷(30℃以下)。会有少量二氯甲烷存在,加入甲醇打浆30分钟,离心,离心固体确定无机械杂质,固体取样测核磁(见图1)、质谱(见图2)、gc和gpc,要求gc大于99%,gpc大于99%(gc:99.7%,gpc:99.5%),收率为72.47%,若不合格,再用重蒸甲醇打浆一小时,降温0-5℃,离心。
[0096]
实施例2
[0097]
将400kg二聚环戊二烯倒入反应釜中,常压内温200℃左右,馏分36~42℃,接收装置为冰浴冷却,收集到包含1,3-环戊二烯的第一料液。
[0098]
向500l釜内抽入丙烯酸,1,3-环戊二烯与丙烯酸的摩尔比为1:1,升温至釜内温度40
±
2℃。釜内氮气保护下,开始滴加第一料液。约14小时滴加完毕。第一料液滴加完毕后,釜内控温40
±
2℃,保温反应6小时,保温结束后取样测gc,要求产品纯度大于90%。反应结束后,循环水降温至25
±
5℃,得到第二料液。碱化处理:将第二料液转入1000l釜内,向釜内抽入甲基叔丁基醚(第二料液与甲基叔丁基醚的质量比为5:1),搅拌10分钟。提前向500l釜内加入水400kg,循环水降温下,缓慢投入氢氧化钠140kg,搅拌溶清,抽入1000l高位槽内。料液釜内控温25
±
5℃,滴加氢氧化钠水溶液,约滴加300l后,釜内搅拌10分钟,取样测ph
值,要求ph为12-13合格,ph值调好后,搅拌10分钟,复测ph。ph为12-13时,开始搅拌30分钟,静止1小时,分层(产品位于水层内)。有机层装桶,水层送样测gc,要求杂质峰小于1%为合格。检测合格后,水层入1000l釜内,有机层入500l釜内,常压回收甲基叔丁基醚。酸化处理:向1000l釜高位槽内抽入盐酸300kg,釜内料液控温25
±
2℃,滴加高位槽内盐酸,调ph为2-3,滴加约250kg后,开始取样测ph,若不合格每滴加20kg盐酸后检测一次,至合格。釜内料液ph值调好后,向釜内抽入甲基叔丁基醚(产品位于有机层内),所述水层相与甲基叔丁基醚的质量比为3:1,搅拌30分钟,静止30分钟,分层,下层水层入另一个1000l釜内,有机层保留在釜内。有机层控温40
±
5℃,减压浓缩甲基叔丁基醚(回收甲基叔丁基醚可以套用)浓缩至无液体流出后,再继续浓缩3小时,取样测gc,要求溶剂残留小于2%,其中浓缩的压力为40mmhg。浓缩到最后料液为油状物,产出油状物325kg。
[0099]
向1000l釜内抽入甲酸,抽入油状物,式i-4化合物与甲酸的摩尔比为1:2,向1000l釜高位槽抽入双氧水,式i-4化合物与双氧水的摩尔比为1:2。滴加双氧水,约8小时滴加完毕。滴加结束后,控温60
±
3℃,保温反应1小时,保温结束后取样测gc,要求原料小于0.5%。提前在1000l釜内抽入水350kg,搅拌下投入亚硫酸钠110kg,搅拌30分钟后,釜内降温5
±
3℃。釜内控温在5
±
5℃将反应合格的料液转入釜内,约8小时转完。釜内控温15
±
3℃,滴加盐酸调ph值为2-3,ph值调好后,料液称重。使用乙酸乙酯进行萃取多次,分层,有机层合并,水层确定无料后放掉。有机层控温40
±
3℃,减压浓缩至无液体流出,再继续浓缩2小时,浓缩压力为45mmhg,取样测gc,要求溶剂残留小于2%。浓缩结束后降温至常温,釜内料液装入干净桶中称重。产出油状物:425kg。向1000l釜内抽入甲醇,抽入油状物,投入碳酸钾,式i-5化合物与甲醇的摩尔比为1:2,与碳酸钾的摩尔比为1:0.2,控温20
±
5℃,搅拌3小时,取样测gc,要求中间态小于5%(中间态位置为主峰后面第一个峰),若不合格每1小时,取样一次至合格为至(反应3小时,中间态为6.02%,反应4小时中间态为4.69%)。反应合格为,釜内控温40
±
5℃,减压浓缩至无液体流出,浓缩压力为20mmhg。釜内浓缩结束后,使用乙酸乙酯进行萃取多次,分层,有机层合并,水层确定无料后放掉。向有机层釜内加入无水硫酸钠25kg,室温搅拌2小时,取样测水份,要求水份小于0.5%。合格为过滤,滤饼用ea20kg淋洗后,过滤,过完后。有机层控温40
±
2℃,减压浓缩至无液体流出,浓缩压力为20mmhg,浓缩结束后釜内有大量白色固体析出,收率为58.1%。
[0100]
向1000l釜内抽入料液、抽入二氯甲烷、抽入三乙胺入釜,式i-6化合物与二氯甲烷的摩尔比为1:5,与三乙胺的摩尔比为1:2,抽料结束后,氮气置换,氮气保护下投入阻聚剂吩噻嗪,式i-6化合物与所述阻聚剂的摩尔比为1:0.01。氮气保护下,釜内控温5
±
2℃开始,滴加甲基丙烯酰氯,式i-6化合物与式i-7化合物的摩尔比为1:1.5,约8小时滴加完毕。滴加完后5
±
2℃,保温反应2小时,取样测gc,要求原料小于0.5%。提前向1000l釜内加入水400kg,降温至2
±
2℃,釜内控温在15℃以下,将反应液转入水解釜内,约3小时转完。转完料后,室温搅拌30分钟,静止30分钟,分层,下层有机层分入周转桶内,水层加入二氯甲烷200kg。室温下搅拌30分钟,静置30分钟,分层,水层确定无料后,放掉,有机层合并,加入水300kg,室温搅拌30分钟,静置30分钟,分层,水层确定无料后,放掉。有机层加入无水硫酸钠50kg,室温搅拌2小时,取样测水份,要求水份小于0.5%过滤,过滤滤饼用二氯甲烷30kg,淋洗,过滤。减压除去二氯甲烷(30℃以下)。会有少量二氯甲烷存在,加入甲醇打浆30分钟,离心,离心固体确定无机械杂质,gc和gpc,要求gc大于99%,gpc大于99%(gc:99.5%,gpc:
99.5%),收率为71.3%,若不合格,再用重蒸甲醇打浆一小时,降温0-5℃,离心。
[0101]
实施例3
[0102]
将400kg二聚环戊二烯倒入反应釜中,常压内温150℃左右,馏分36~42℃,接收装置为冰浴冷却,收集到包含1,3-环戊二烯的第一料液。
[0103]
向500l釜内抽入丙烯酸,1,3-环戊二烯与丙烯酸的摩尔比为1.5:1,升温至釜内温度40
±
2℃。釜内氮气保护下,开始滴加第一料液。约14小时滴加完毕。第一料液滴加完毕后,釜内控温40
±
2℃,保温反应6小时,保温结束后取样测gc,要求产品纯度大于90%。反应结束后,循环水降温至25
±
5℃,得到第二料液。碱化处理:将第二料液转入1000l釜内,向釜内抽入甲基叔丁基醚(第二料液与甲基叔丁基醚的质量比为2:1),搅拌10分钟。提前向500l釜内加入水400kg,循环水降温下,缓慢投入氢氧化钠140kg,搅拌溶清,抽入1000l高位槽内。料液釜内控温25
±
5℃,滴加氢氧化钠水溶液,约滴加300l后,釜内搅拌10分钟,取样测ph值,要求ph为12-13合格,ph值调好后,搅拌10分钟,复测ph。ph为12-13时,开始搅拌30分钟,静止1小时,分层(产品位于水层内)。有机层装桶,水层送样测gc,要求杂质峰小于1%为合格。检测合格后,水层入1000l釜内,有机层入500l釜内,常压回收甲基叔丁基醚。酸化处理:向1000l釜高位槽内抽入盐酸300kg,釜内料液控温25
±
2℃,滴加高位槽内盐酸,调ph为2-3,滴加约250kg后,开始取样测ph,若不合格每滴加20kg盐酸后检测一次,至合格。釜内料液ph值调好后,向釜内抽入甲基叔丁基醚(产品位于有机层内),所述水层相与甲基叔丁基醚的质量比为1:1,搅拌30分钟,静止30分钟,分层,下层水层入另一个1000l釜内,有机层保留在釜内。有机层控温40
±
5℃,减压浓缩甲基叔丁基醚(回收甲基叔丁基醚可以套用)浓缩至无液体流出后,再继续浓缩3小时,取样测gc,要求溶剂残留小于2%,其中浓缩的压力为50mmhg。浓缩到最后料液为油状物,产出油状物315kg。
[0104]
向1000l釜内抽入甲酸,抽入油状物,式i-4化合物与甲酸的摩尔比为1:1,向1000l釜高位槽抽入双氧水,式i-4化合物与双氧水的摩尔比为1:1。滴加双氧水,约8小时滴加完毕。滴加结束后,控温60
±
3℃,保温反应1小时,保温结束后取样测gc,要求原料小于0.5%。提前在1000l釜内抽入水350kg,搅拌下投入亚硫酸钠110kg,搅拌30分钟后,釜内降温5
±
3℃。釜内控温在5
±
5℃将反应合格的料液转入釜内,约8小时转完。釜内控温15
±
3℃,滴加盐酸调ph值为2-3,ph值调好后,料液称重。使用乙酸乙酯进行萃取多次,分层,有机层合并,水层确定无料后放掉。有机层控温40
±
3℃,减压浓缩至无液体流出,再继续浓缩2小时,浓缩压力为50mmhg,取样测gc,要求溶剂残留小于2%。浓缩结束后降温至常温,釜内料液装入干净桶中称重。产出油状物:421kg。
[0105]
向1000l釜内抽入甲醇,抽入油状物,投入碳酸钾,式i-5化合物与甲醇的摩尔比为1:5,与碳酸钾的摩尔比为1:1,控温20
±
5℃,搅拌3小时,取样测gc,要求中间态小于5%(中间态位置为主峰后面第一个峰),若不合格每1小时,取样一次至合格为至(反应3小时,中间态为6.02%,反应4小时中间态为4.69%)。反应合格为,釜内控温40
±
5℃,减压浓缩至无液体流出,浓缩压力为50mmhg。釜内浓缩结束后,使用乙酸乙酯进行萃取多次,分层,有机层合并,水层确定无料后放掉。向有机层釜内加入无水硫酸钠25kg,室温搅拌2小时,取样测水份,要求水份小于0.5%。合格为过滤,滤饼用ea20kg淋洗后,过滤,过完后。有机层控温40
±
2℃,减压浓缩至无液体流出,浓缩压力为50mmhg,浓缩结束后釜内有大量白色固体析出,收率为57.1%。
[0106]
向1000l釜内抽入料液、抽入二氯甲烷、抽入三乙胺入釜,式i-6化合物与二氯甲烷的摩尔比为1:10,与三乙胺的摩尔比为1:1.5,抽料结束后,氮气置换,氮气保护下投入阻聚剂吩噻嗪,式i-6化合物与所述阻聚剂的摩尔比为1:0.05。氮气保护下,釜内控温5
±
2℃开始,滴加甲基丙烯酰氯,式i-6化合物与式i-7化合物的摩尔比为1:1,约8小时滴加完毕。滴加完后5
±
2℃,保温反应2小时,取样测gc,要求原料小于0.5%。提前向1000l釜内加入水400kg,降温至2
±
2℃,釜内控温在15℃以下,将反应液转入水解釜内,约3小时转完。转完料后,室温搅拌30分钟,静止30分钟,分层,下层有机层分入周转桶内,水层加入二氯甲烷200kg。室温下搅拌30分钟,静置30分钟,分层,水层确定无料后,放掉,有机层合并,加入水300kg,室温搅拌30分钟,静置30分钟,分层,水层确定无料后,放掉。有机层加入无水硫酸钠50kg,室温搅拌2小时,取样测水份,要求水份小于0.5%过滤,过滤滤饼用二氯甲烷30kg,淋洗,过滤。减压除去二氯甲烷(30℃以下)。会有少量二氯甲烷存在,加入甲醇打浆30分钟,离心,离心固体确定无机械杂质,gc和gpc,要求gc大于99%,gpc大于99%(gc:99.56%,gpc:99.6%),收率为70.6%,若不合格,再用重蒸甲醇打浆一小时,降温0-5℃,离心。
[0107]
实施例4
[0108][0109]
将400kg式i-1化合物倒入反应釜中,常压内温165℃左右,馏分36~42℃,接收装置为冰浴冷却,收集到包含式i-2化合物的第一料液。
[0110]
向500l釜内抽入式i-3化合物,式i-2化合物与式i-3化合物的摩尔比为1.2:1,升温至釜内温度40
±
2℃。釜内氮气保护下,开始滴加第一料液。约14小时滴加完毕。第一料液滴加完毕后,釜内控温40
±
2℃,保温反应6小时,保温结束后取样测gc,要求产品纯度大于90%。反应结束后,循环水降温至25
±
5℃,得到第二料液。碱化处理:将第二料液转入1000l釜内,向釜内抽入甲基叔丁基醚(第二料液与甲基叔丁基醚的质量比为4:1),搅拌10分钟。提前向500l釜内加入水400kg,循环水降温下,缓慢投入氢氧化钠140kg,搅拌溶清,抽入1000l高位槽内。料液釜内控温25
±
5℃,滴加氢氧化钠水溶液,约滴加300l后,釜内搅拌10分钟,取样测ph值,要求ph为12-13合格,ph值调好后,搅拌10分钟,复测ph。ph为12-13时,开始搅拌30分钟,静止1小时,分层(产品位于水层内)。有机层装桶,水层送样测gc,要求杂质峰小于1%为合格。检测合格后,水层入1000l釜内,有机层入500l釜内,常压回收甲基叔丁基醚。酸化处理:向1000l釜高位槽内抽入盐酸300kg,釜内料液控温25
±
2℃,滴加高位槽内盐酸,调ph为2-3,滴加约250kg后,开始取样测ph,若不合格每滴加20kg盐酸后检测一次,至合格。釜内料液ph值调好后,向釜内抽入甲基叔丁基醚(产品位于有机层内),所述水层相与甲基叔丁基醚的质量比为2:1,搅拌30分钟,静止30分钟,分层,下层水层入另一个1000l釜内,有机层保留在釜内。有机层控温40
±
5℃,减压浓缩甲基叔丁基醚(回收甲基叔丁基醚可以套用)浓缩至无液体流出后,再继续浓缩3小时,取样测gc,要求溶剂残留小于2%,其中浓
缩的压力为30mmhg。浓缩到最后料液为油状物,产出式i-4化合物的油状物330kg。
[0111]
向1000l釜内抽入甲酸,抽入式i-4化合物的油状物,式i-4化合物与甲酸的摩尔比为1:1.5,向1000l釜高位槽抽入过氧乙酸,式i-4化合物与过氧乙酸的摩尔比为1:2。滴加双氧水,约8小时滴加完毕。滴加结束后,控温60
±
3℃,保温反应1小时,保温结束后取样测gc,要求原料小于0.5%。提前在1000l釜内抽入水350kg,搅拌下投入亚硫酸钠110kg,搅拌30分钟后,釜内降温5
±
3℃。釜内控温在5
±
5℃将反应合格的料液转入釜内,约8小时转完。釜内控温15
±
3℃,滴加盐酸调ph值为2-3,ph值调好后,料液称重。使用乙酸乙酯进行萃取多次,分层,有机层合并,水层确定无料后放掉。有机层控温40
±
3℃,减压浓缩至无液体流出,再继续浓缩2小时,浓缩压力为20mmhg,取样测gc,要求溶剂残留小于2%。浓缩结束后降温至常温,釜内料液装入干净桶中称重。产出式i-5化合物的油状物:490kg。
[0112]
向1000l釜内抽入甲醇,抽入油状物,投入碳酸钾,式i-5化合物与甲醇的摩尔比为1:3,与碳酸钾的摩尔比为1:0.6,控温20
±
5℃,搅拌3小时,取样测gc,要求中间态小于5%(中间态位置为主峰后面第一个峰),若不合格每1小时,取样一次至合格为至(反应3小时,中间态为6.02%,反应4小时中间态为4.69%)。反应合格为,釜内控温40
±
5℃,减压浓缩至无液体流出,浓缩压力为30mmhg。釜内浓缩结束后,使用乙酸乙酯进行萃取多次,分层,有机层合并,水层确定无料后放掉。向有机层釜内加入无水硫酸钠25kg,室温搅拌2小时,取样测水份,要求水份小于0.5%。合格为过滤,滤饼用ea20kg淋洗后,过滤,过完后。有机层控温40
±
2℃,减压浓缩至无液体流出,浓缩压力为30mmhg,浓缩结束后釜内有大量白色固体析出,收率为59.2%。
[0113]
向1000l釜内抽入料液、抽入二氯甲烷、抽入三乙胺入釜,式i-6化合物与二氯甲烷的摩尔比为1:7,与三乙胺的摩尔比为1:1.8,抽料结束后,氮气置换,氮气保护下投入阻聚剂吩噻嗪,式i-6化合物与所述阻聚剂的摩尔比为1:0.03。氮气保护下,釜内控温5
±
2℃开始,滴加式i-7化合物丙烯酰氯,式i-6化合物与式i-7化合物的摩尔比为1:1.2,约8小时滴加完毕。滴加完后5
±
2℃,保温反应2小时,取样测gc,要求原料小于0.5%。提前向1000l釜内加入水400kg,降温至2
±
2℃,釜内控温在15℃以下,将反应液转入水解釜内,约3小时转完。转完料后,室温搅拌30分钟,静止30分钟,分层,下层有机层分入周转桶内,水层加入二氯甲烷200kg。室温下搅拌30分钟,静置30分钟,分层,水层确定无料后,放掉,有机层合并,加入水300kg,室温搅拌30分钟,静置30分钟,分层,水层确定无料后,放掉。有机层加入无水硫酸钠50kg,室温搅拌2小时,取样测水份,要求水份小于0.5%过滤,过滤滤饼用二氯甲烷30kg,淋洗,过滤。减压除去二氯甲烷(30℃以下)。会有少量二氯甲烷存在,加入甲醇打浆30分钟,离心,离心式i化合物固体确定无机械杂质,gc和gpc,要求gc大于99%,gpc大于99%(gc:99.4%,gpc:99.5%),收率为70.8%,若不合格,再用重蒸甲醇打浆一小时,降温0-5℃,离心。
[0114]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1