一步法合成2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的方法
1.本发明涉及一步法合成2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的方法,属于有机化合物的制备领域。
背景技术:
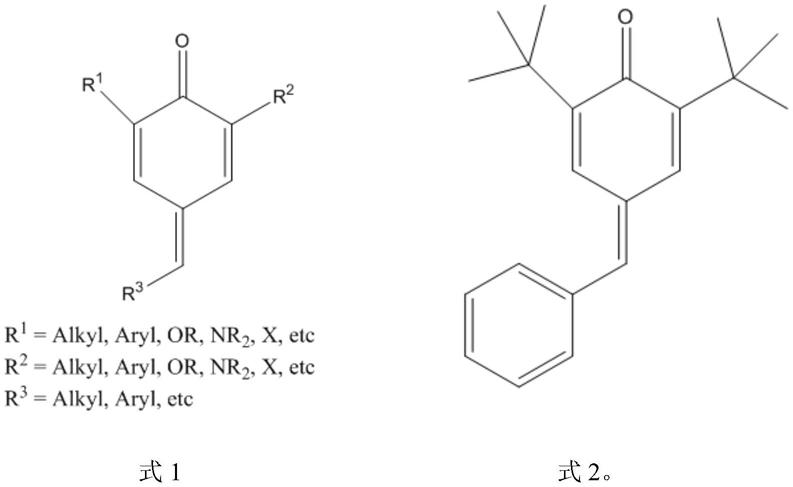
2.2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮属于对亚甲基苯醌类化合物(式1),因其独特的骨架结构和特殊的亲电活性,具有更容易发生亲核加成-芳构化反应而构建一系列不同结构、不同类型化合物的性能,使得此类反应底物受到科研工作者们的青睐,在有机合成领域有着举足轻重的地位。同时,2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮也可以作为5-脂氧合酶和环氧酶的酶抑制剂,可以应用于多种疾病的治疗,也可以应用于抗氧剂和阻聚剂,其主要用于医药、农药和精细化工等领域。其中,2,6-二叔丁基-4-苯亚甲基-2,5-环己二烯基-1-酮是典型代表,其具体的结构为式2所示:
[0003][0004]
至今,已有很多文献报道对亚甲基苯醌类化合物在有机合成领域中的应用。如,以对亚甲基苯醌类化合物为底物,在无溶剂条件下碳酸铯催化叠氮芳香化反应得到定量收率的苄基叠氮化合物(eur.j.org.chem.2020,6068);又如,以对亚甲基苯醌类化合物为底物,无溶剂条件下碳酸铯催化硅烷化反应高收率地得到苄基硅烷类化合物(rsc,adv.,2021,17860);再如,以对亚甲基苯醌类化合物为底物,铜催化不对称1,6-硼加成反应高收率、高对映选择性地得到苄基硼酸酯类化合物(angew.chem.int.ed.,2015,12134;acs catal.2016,6,442);也可以作为一种抗氧剂和阻聚剂应用于聚合物的制备过程(us2008132726a1)。由此看来,对亚甲基苯醌类化合物的简单高效合成,是对亚甲基苯醌类化合物作为反应底物应用于有机合成领域和精细化工领域的重要前提。
[0005]
目前,现有技术中已有对亚甲基苯醌类化合物合成方法的报道。如,美国专利文献
us4032547公开了一种新的对亚甲基苯醌类化合物的合成工艺方法,该方法采用在氢氧化钠、铁氰化钠的水溶液与2,6-二叔丁基-4-乙基苯酚溶于己烷的混合液中,搅拌下1.8小时内滴加过硫酸钠的水溶液,控制反应温度在27-33℃并继续搅拌反应4小时,分液、干燥、蒸出溶剂且重结晶后得到2,6-二叔丁基-4-乙烯基-2,5-环己二烯基酮,最高收率可达92%;但该发明采用氢氧化钠、铁氰化钠的水溶液为催化剂,过硫酸钠为氧化剂、2,6-二叔丁基-4-乙基苯酚为原料的催化氧化方法,所用原料不易得、成本较高,且废水量大;又如,中国专利文献cn105418395a公开了一种2,6-二叔丁基-4-苯亚甲基-2,5-环己二烯基-1-酮的制备方法,该方法采用六氢吡啶作为缩合剂,2,6-二叔丁基苯酚和苯甲醛在甲苯溶剂中回流分水3小时以上,反应先生成中间体曼尼希碱,再加入醋酸酐消去反应,加甲苯,水洗,饱和食盐水洗,无水硫酸钠干燥,过滤,减压蒸出溶剂,混合溶剂(乙酸乙酯:正己烷体积比=3:1)重结晶,得2,6-二叔丁基-4-苯亚甲基-2,5-环己二烯基-1-酮,收率:79.1%;该发明实际上是一锅两步反应,即先生成曼尼希碱,曼尼希碱不分离直接和后加入的乙酸酐发生消去反应;因此,要消耗化学计量以上的六氢吡啶和醋酸酐,同时产生等量的n-乙酰基六氢吡啶副产物,工艺繁琐,成本较高。
[0006]
因此,亟需一种原料种类少,成本较低,所使用的原料廉价、易得,产品收率高,一步反应制备2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的方法。
技术实现要素:
[0007]
针对现有技术中存在的不足,本发明提供一步法合成2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的方法。
[0008]
本发明的方法仅使用催化剂吡咯烷,吡咯烷和芳香基甲醛缩合得到亚胺盐,亚胺盐作为亲电试剂进攻富电子的2,6-二取代苯酚进而得到2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮,无曼尼希碱生成,也无需消去反应处理步骤,一步反应得到终产品。所使用的原料廉价、易得,合成操作步骤简单、温和,无大量废水和副产物,而且能够获得比较好的收率和高纯度,适合大量制备和规模化生产。
[0009]
本发明是通过如下技术方案实现的:
[0010]
一步法合成2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的方法,步骤如下:
[0011]
将2,6-二取代苯酚、芳香基甲醛和催化剂吡咯烷溶于有机溶剂中,得混合液,搅拌反应1-72小时,得反应液;反应液回收溶剂和吡咯烷后,得粗产物,粗产物重结晶提纯,得到2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮。
[0012]
根据本发明优选的,2,6-二取代苯酚的结构如下式ⅰ所示:
[0013]
[0014]
其中r1、r2为烷基、环烷基、芳香基、杂环基、烷氧基、芳氧基、卤素、烷氨基或芳氨基。
[0015]
根据本发明优选的,烷基为1-20个碳原子的烷基,环烷基为3-10个碳原子的环烷基,芳香基为6-30个碳原子的芳香基,杂环基为2-30个碳原子的杂环基,烷氧基为1-20个碳原子的烷氧基,芳氧基为6-30个碳原子的芳氧基,烷氨基为1-40个碳原子的烷氨基,芳氨基为6-30个碳原子的芳氨基。
[0016]
根据本发明优选的,芳香基甲醛的结构如下式ⅱ所示:
[0017][0018]
其中r3为芳香烃基或芳杂环基。
[0019]
根据本发明优选的,芳香烃基为6-30个碳原子的芳香烃基,芳杂环基为4-30个碳原子的芳杂环基。
[0020]
根据本发明优选的,吡咯烷的分子结构式如下式ⅲ所示:
[0021][0022]
根据本发明优选的,所述有机溶剂为醇类、烃类、醚类、酯类或卤代烃类溶剂。
[0023]
进一步优选的,所述有机溶剂为甲醇、乙醇、丙醇、异丙醇、丁醇、二氯甲烷或乙酸乙酯。
[0024]
最为优选的,所述有机溶剂为甲醇、乙醇、丙醇、异丙醇或丁醇。
[0025]
根据本发明优选的,2,6-二取代苯酚与芳香基甲醛的摩尔比为1:0.1~10.0。
[0026]
进一步优选的,2,6-二取代苯酚与芳香基甲醛的摩尔比为1:0.5~5。
[0027]
最为优选的,2,6-二取代苯酚与芳香基甲醛的摩尔比为1:1~1.1。
[0028]
根据本发明优选的,2,6-二取代苯酚与吡咯烷的摩尔比为1:0.1~10.0。
[0029]
进一步优选的,2,6-二取代苯酚与吡咯烷的摩尔比为1:0.5~5。
[0030]
最为优选的,2,6-二取代苯酚与吡咯烷的摩尔比为1:1~1.1。
[0031]
根据本发明优选的,混合液中2,6-二取代苯酚的浓度为0.005~0.5g/ml。
[0032]
进一步优选的,混合液中2,6-二取代苯酚的浓度为0.05~0.3g/ml。
[0033]
最为优选的,混合液中2,6-二取代苯酚的浓度为0.1~0.3g/ml。
[0034]
根据本发明优选的,反应温度为-35~80℃,更优选的反应温度为0~60℃,最优选的反应温度为25~50℃。
[0035]
根据本发明优选的,反应过程中通过薄层色谱跟踪检测反应进程,当2,6-二取代苯酚完全消耗,停止反应。
[0036]
根据本发明优选的,通过常压蒸馏或减压旋蒸回收溶剂和吡咯烷,旋蒸条件为:旋蒸压力为:0.00~-0.095mpa,旋蒸温度为25~90℃。
[0037]
更优选的旋蒸压力为:-0.030~-0.090mpa,旋蒸温度为35~70℃;最优选的旋蒸压力为:-0.050~-0.070mpa,旋蒸温度为45~65℃。
[0038]
根据本发明优选的,重结晶提纯是按如下方法进行的:
[0039]
将粗产物在加热回流的情况下溶解在醇类或醇-水或石油醚或石油醚-乙酸乙酯混合溶剂中,70~100℃加热回流10~20分钟,自然冷却到室温,在室温下静置,使其慢慢析出,得到重结晶后的纯品;粗产物与溶剂的质量体积比为1:(3-10),单位g/ml。
[0040]
本发明的技术特点及有益效果:
[0041]
本发明的合成路线如下式ⅳ所示:
[0042][0043]
2,6-二取代苯酚和芳香基甲醛溶于有机溶剂中,在催化剂吡咯烷存在的情况下,吡咯烷和芳香基甲醛缩合得到亚胺盐,亚胺盐作为亲电试剂进攻富电子的2,6-二取代苯酚进而直接得到2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮,同时又再生催化剂吡咯烷,吡咯烷做催化剂,中间无曼尼希碱生成,也无需消去反应处理步骤,通过薄层色谱跟踪检测反应进程,当2,6-二取代苯酚消耗殆尽,反应停止得反应液;将反应液经蒸馏或减压旋蒸回收溶剂和吡咯烷(回收的溶剂和吡咯烷可以循环使用),得粗产物;粗产物通过重结晶的方法进行提纯,得到含量大于98%的2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮。
[0044]
本发明的优点如下:
[0045]
1、本发明仅使用催化剂吡咯烷,不需要乙酸酐或其他酸性催化剂和助剂,也不需要氧化剂等无机化学品,也不必需高温、高真空蒸馏步骤;原料种类少,成本较低,所使用的原料廉价、易得;制备操作步骤简单温和,无大量废水和副产物,而且能够获得比较好的收率和高纯度,适合2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的大量制备和规模化生产。
[0046]
2、本发明的合成方法能够获得比较高的收率和高的化学纯度;2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的总收率高达91%,含量大于98%。
具体实施方式
[0047]
下面结合具体实施例对本发明做进一步的说明,但不限于此。
[0048]
同时下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材
料,如无特殊说明,均可从商业途径获得。
[0049]
本发明涉及的总收率是指重结晶达标产品加上重结晶母液回收的粗产品再多次重结晶达标产品之和/理论产量。
[0050]
实施例1
[0051]
一步法合成2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的方法,步骤如下:
[0052]
(1)装有电动搅拌装置的2升三口烧瓶中,加入2,6-二叔丁基苯酚206.3克(1.0mol)、苯甲醛112毫升(116.7克,1.1mol)和吡咯烷83毫升(71.1克,1.0mol),再加入甲醇1000毫升,25℃下搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基苯酚完全消耗,停止反应(约需12小时),常压蒸馏回收甲醇和吡咯烷,得粗产物的蜡状固体;
[0053]
(2)将粗产物的蜡状固体90℃加热回流条件下溶解在1500ml石油醚中,加热回流10分钟,自然冷却到室温,在室温下静置,使其慢慢析出;必要时可重复上述操作,最终得到重结晶后的纯品257g(收率87%,含量98.7%)。熔点为:75-77℃,核磁分析为:1h nmr(500mhz,cdcl3,rt)δ7.58(d,j=2.4hz,1h),7.54-7.47(m,3h)7.44-7.39(m,2h),7.24(s,1h),7.08(d,j=2.4,1h),1.36(s,9h),1.34(s,9h).
[0054]
实施例2
[0055]
一步法合成2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的方法,步骤如下:
[0056]
(1)装有电动搅拌装置的2升三口烧瓶中,加入2,6-二叔丁基苯酚206.3克(1.0mol)、苯甲醛112毫升(116.7克,1.1mol)和吡咯烷83毫升(71.1克,1.0mol),再加入乙醇1000毫升,25℃下搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基苯酚完全消耗,停止反应(约需25小时),减压旋蒸以回收乙醇和吡咯烷,得粗产物的蜡状固体,所述旋蒸条件为:旋蒸压力为:-0.090mpa,旋蒸温度为40-60℃。
[0057]
(2)将粗产物的蜡状固体90℃加热回流条件下溶解在1000ml石油醚中,加热回流10分钟,自然冷却到室温,在室温下静置,使其慢慢析出;必要时可重复上述操作,最终得到重结晶后的纯品183g(收率62%,含量98.4%)。
[0058]
实施例3
[0059]
一步法合成2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的方法,步骤如下:
[0060]
(1)装有电动搅拌装置的2升三口烧瓶中,加入2,6-二叔丁基苯酚206.3克(1.0mol)、苯甲醛112毫升(116.7克,1.1mol)和吡咯烷41.5毫升(35.5克,0.5mol),再加入甲醇1000毫升,25℃下搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基苯酚完全消耗,停止反应(约需48小时),常压蒸馏以回收甲醇和吡咯烷,得粗产物的蜡状固体。
[0061]
(2)将粗产物的蜡状固体90℃加热回流条件下溶解在1500ml石油醚中,加热回流10分钟,自然冷却到室温,在室温下静置,使其慢慢析出;必要时可重复上述操作,最终得到重结晶后的纯品248g(收率84%,含量98.6%)。
[0062]
实施例4
[0063]
一步法合成2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的方法,步骤如下:
[0064]
(1)装有电动搅拌装置的2升三口烧瓶中,加入2,6-二叔丁基苯酚206.3克(1.0mol)、4-甲氧基苯甲醛134毫升(149.8克,1.1mol)和吡咯烷83毫升(71.1克,1.0mol),再加入甲醇1000毫升,25℃下搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基苯酚完全消耗,停止反应(约需10小时),常压蒸馏以回收甲醇和吡咯烷,得粗产物的蜡状固体。
[0065]
(2)将粗产物的蜡状固体90℃加热回流条件下溶解在1700ml石油醚中,加热回流10分钟,自然冷却到室温,在室温下静置,使其慢慢析出;必要时可重复上述操作,最终得到重结晶后的纯品270g(收率83%,含量98.8%)。熔点为:127-128℃,核磁分析为:1h nmr(500mhz,cdcl3,rt)δ7.57(d,j=2.4hz,1h),7.45(d,j=8.7hz,2h)7.14(s,1h),7.02-6.97(m,3h),3.88(s,3h),1.34(s,9h),1.33(s,9h).
[0066]
实施例5
[0067]
一步法合成2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的方法,步骤如下:
[0068]
(1)装有电动搅拌装置的2升三口烧瓶中,加入2,6-二叔丁基苯酚206.3克(1.0mol)、4-三氟甲基苯甲醛148.5毫升(191.5克,1.1mol)和吡咯烷83毫升(71.1克,1.0mol),再加入甲醇1000毫升,25℃下搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基苯酚完全消耗,停止反应(约需24小时),常压蒸馏以回收甲醇和吡咯烷,得粗产物的蜡状固体。
[0069]
(2)将粗产物的蜡状固体90℃加热回流条件下溶解在1200ml石油醚中,加热回流10分钟,自然冷却到室温,在室温下静置,使其慢慢析出;必要时可重复上述操作,最终得到重结晶后的纯品240g(收率66%,含量98.5%)。熔点为:125-127℃,核磁分析为:1h nmr(500mhz,cdcl3,rt)δ7.72(d,j=8.2hz,2h),7.56(d,j=8.2hz,2h)7.43(d,j=2.4hz,1h),7.17(s,1h),7.03(d,j=2.4,1h),1.34(s,9h),1.30(s,9h).
[0070]
实施例6
[0071]
一步法合成2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的方法,步骤如下:
[0072]
(1)装有电动搅拌装置的2升三口烧瓶中,加入2,6-二叔丁基苯酚206.3克(1.0mol)、3-吡啶甲醛103毫升(117.8克,1.1mol)和吡咯烷83毫升(71.1克,1.0mol),再加入甲醇1000毫升,25℃下搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基苯酚完全消耗,停止反应(约需24小时),常压蒸馏以回收甲醇和吡咯烷,得粗产物的蜡状固体。
[0073]
(2)将粗产物的蜡状固体90℃加热回流条件下溶解在2100ml石油醚中,加热回流10分钟,自然冷却到室温,在室温下静置,使其慢慢析出;必要时可重复上述操作,最终得到重结晶后的纯品237g(收率80%,含量98.3%)。熔点为:81-83℃,核磁分析为:1h nmr(500mhz,cdcl3,rt)δ8.72(d,j=1.3hz,1h),8.62(dd,j=4.7hz,j=1.1hz,1h),7.78(d,j=8.0hz,1h),7.46-7.36(m,2h),7.12(s,1h),7.04(d,j=2.4,1h),1.35(s,9h),1.31(s,9h).
[0074]
实施例7
[0075]
一步法合成2,6-二取代基-4-芳香基亚甲基-2,5-环己二烯-1-酮的方法,步骤如
下:
[0076]
(1)装有电动搅拌装置的2升三口烧瓶中,加入2,6-二叔丁基苯酚206.3克(1.0mol)、1-萘甲醛171.8克(1.1mol)和吡咯烷83毫升(71.1克,1.0mol),再加入甲醇1000毫升,25℃下搅拌反应且通过薄层色谱跟踪检测反应进程;当2,6-二叔丁基苯酚完全消耗,停止反应(约需12小时),常压蒸馏以回收甲醇和吡咯烷,得粗产物的蜡状固体。
[0077]
(2)将粗产物的蜡状固体90℃加热回流条件下溶解在1900ml石油醚中,加热回流10分钟,自然冷却到室温,在室温下静置,使其慢慢析出;必要时可重复上述操作,最终得到重结晶后的纯品314g(收率91%,含量98.7%)。熔点为:139-140℃。
[0078]
对产品进行核磁鉴定,核磁数据如下:1h nmr(500mhz,cdcl3,rt)δ8.08-8.01(m,1h),7.96-7.91(m,2h),7.79(s,1h),7.62-7.54(m,3h),7.49(d,j=7.1hz,1h),7.41(d,j=2.2hz,1h),7.22(d,j=2.2,1h),1.40(s,9h),1.27(s,9h).
[0079]
对比例1
[0080]
同实施例1所述的制备方法,不同之处在于:
[0081]
步骤(1)中,用四氢呋喃替换甲醇,其他按实施例1进行,
[0082]
步骤(2)中,将粗产物的蜡状固体90℃加热回流条件下溶解在1500ml石油醚中,加热回流10分钟,自然冷却到室温,在室温下静置,使其慢慢析出;必要时可重复上述操作,得到重结晶后的纯品162g(收率55%,含量98.2%)。
[0083]
对比例2
[0084]
同实施例1所述的制备方法,不同之处在于:
[0085]
步骤(1)中,用二氯乙烷替换甲醇,其他按实施例1进行,
[0086]
步骤(2)中,将粗产物的蜡状固体90℃加热回流条件下溶解在1500ml石油醚中,加热回流10分钟,自然冷却到室温,在室温下静置,使其慢慢析出;必要时可重复上述操作。得到重结晶后的纯品127g(收率43%,含量98.2%)。
[0087]
对比例3
[0088]
同实施例1所述的制备方法,不同之处在于:
[0089]
步骤(1)中,用甲苯替换甲醇,其他按实施例1进行,
[0090]
步骤(2)中,将粗产物的蜡状固体90℃加热回流条件下溶解在1500ml石油醚中,加热回流10分钟,自然冷却到室温,在室温下静置,使其慢慢析出;必要时可重复上述操作,得到重结晶后的纯品106g(收率36%,含量98.3%)。
[0091]
通过对比例1-3可以看出,采用四氢呋喃、二氯乙烷、甲苯做溶剂,收率明显小于本发明的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1