一种3-氨基吡咯烷二盐酸盐合成方法与流程
1.本发明涉及3-氨基吡咯烷二盐酸盐合成方法。
背景技术:
2.3-氨基吡咯烷二盐酸盐及其光学异构体是合成大量药物的关键中间体。通过是旋光3-氨基吡咯二盐酸盐是制备农用化学品如乙烯基吡咯烷酮-头孢菌素衍生物和药学活性物质妥舒沙星和其它喹诺酮类抗菌药的关键中间体。
3.日本公开特许jp61057579a报道:n-乙氧羰基甘氨酸乙酯先对丙烯酸乙酯加成,接着进行dieckmann环合,之后选择性水解并脱羧得吡咯烷酮,吡咯烷酮依次经肟化、还原和酸性水解并脱羧即得3-氨基吡咯烷。
4.日本公开特许jp3133954a报道:n-苄基甘氨酸乙酯先对丙烯酸乙酯加成,接着在t-buok存在下进行dieckmann环合得n-苄基-4-甲酸乙酯基-3-吡咯烷酮,再依次经水解并脱羧得1-苄基-3-吡咯烷酮,还原氨化,并氢解脱苄基即得3-氨基吡咯烷。
5.日本公开的合成路线中,存在着如下问题:1、反应路线长,步骤多,总收率低,污染大;2、某些反应路线涉及成肟反应,导致了更低的收率;3、脱苄基时的反应条件苛刻,高温高压不易控制;3-氨基吡咯烷的工业化生产成本过高。
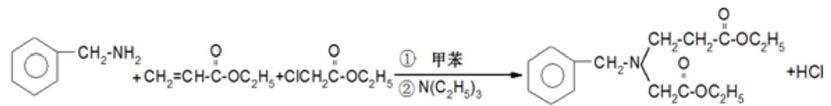
6.中国专利申请cn2006101501116,公开了一种以苄胺、丙烯酸乙酯、氯乙酸乙酯为主要起始原料,通过加成、环和、水解、氢化(两步),制备3-氨基吡咯烷合成方法;避免了肟中间体的生成,且相对日本公开的方法提高了收率,得到了一种新的工艺简单、成本低合成方法;但是由于原料反应活性高,虽然收率提高,但是产品的收率不稳定,出现副产品3-羟基吡咯烷醇、4-氨基哌啶、3-甲氨基吡咯烷,且副品产量不稳定;通过有机溶剂的蒸馏提纯提高产品的纯度,增加了合成成本。
技术实现要素:
7.为了解决上述技术问题,本发明的目的在于提供一种陶瓷表面加工方法:将陶瓷坯体表面划分透波区、屏蔽区;对透波区覆盖保护层进行防护;对屏蔽层进行等离子处理;配制第一金属浆料、第二金属浆料、第三金属浆料;将第一金属浆料附着在屏蔽层表面,形成金属过渡层;将第二金属浆料附着在金属过渡层远离陶瓷坯体的一面,形成导电层;解决石英纤维增强复合材料具部透波率提高,且在使用过程中透波区域透波率不降低。
8.根据本发明提供的一种3-氨基吡咯烷二盐酸盐合成方法,包括以下步骤:
9.1)甲苯、苄胺加入反应容器中;对所述甲苯、苄胺进行降温冷却;
10.反应容器内物料降温后,分次向反应容器内加入丙烯酸乙酯,自然升温;自然升温至温度不再发生变化;
11.然后加入氯化氢气体,搅拌静置;再加入氯乙酸乙酯、三乙胺,升温至第一温度,持续时间为第一时长;
12.将所得的反应溶液进行分离、蒸馏得到苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧
基甲基)胺液;
[0013][0014]
2)甲苯、苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺液加入反应容器中,所述反应容器进行降温冷却;然后加入乙醇钠,控制加料时温度至第二温度,加料完毕控制温度至第三温度,持续时间为第二时间;
[0015]
然后将得到的反应溶液与纯化水、盐酸在反应容器中混合,调节到第一ph;
[0016]
然后将反应液分离、蒸馏,用氢氧化钠水溶液调至第二ph,再用冰醋酸调至第三ph;分离后得到1-苄基-3-吡咯烷酮液;
[0017][0018]
3)甲醇、1-苄基-3-吡咯烷酮液加入反应容器,加入活性镍;然后通入液氨,再对反应容器内物料进行加热;
[0019]
然后向反应容器内通入氢气,进行氢化反应;反应完毕后进行分离、蒸馏得到1-苄基-3-氨基吡咯烷液;
[0020][0021]
4)将得到的1-苄基-3-氨基吡咯烷液加入氯化氢进行成盐反应,然后分离、干燥得到1-苄基-3-氨基吡咯烷盐;
[0022][0023]
5)将甲醇加入高压反应容器中,加入1-苄基-3-氨基吡咯烷盐,然后加入钯炭1kg,纯化水10kg,进行通入氮气置换反应容器内空气后,通入氢气;
[0024]
通入氢气时提高氢压至2-3mpa,并升高温度,反应过程中维持温度为第四温度,进行氢化反应,当时温度上升,压力同时上升反应结束,得到3-氨基吡咯烷液;
[0025][0026]
6)3-氨基吡咯烷液加入反应容器中;所述反应容器进行降温冷却;
[0027]
加入盐酸调节ph为2-3,进行蒸馏、干燥得到3-氨基吡咯烷二盐酸盐。
[0028]
采用上述进一步技术方案的有益效果在于,通过先对甲苯、苄胺混合溶液进行降
温冷却后,加入丙烯酸乙酯,且在加入氯乙酸乙酯、三乙胺之前进行自然升温之平稳,由于丙烯酸乙酯活性高,避免了丙烯酸乙酯与苄胺时反应剧烈,以及避免了丙烯酸乙酯进行自聚合反应产生二聚体或多聚体以及二聚体与苄胺加成反应等副反应,避免了杂质的增加,主要是避免了3-苄胺基丙酸乙酯的产出率,且有利于避免了成品中4-氨基哌啶产生;
[0029]
通过入氯乙酸乙酯、三乙胺之前通入氯化氢,抑制了少量的丙烯酸乙酯与3-苄胺基丙酸乙酯进一步的加成反应产生,进一步避免了产率的降低;
[0030]
通过加入氯乙酸乙酯、三乙胺,缓慢升温至第一温度,解决了氯化氢加入后对苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺合成效率降低问题,且不在反应过程中添加其他杂质或副产品;
[0031]
通过在加入乙醇钠之前进行冷却,降低苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺的活性,避免在乙醇钠加入时,溶液未混合均匀,导致的少量苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺与过量的乙醇钠反应,发生副反应,同时通过在稳步升温的条件下,既实现了环化反应,同时稳定控制反应的活性,进一步提高的产率;通过少量冰醋酸调至第三ph,可以达到更优ph,且形成缓冲体系保持ph稳定,有利于避免了副反应产生1-苄基-3-吡咯烷醇,同时抑制了得到的1-苄基-3-吡咯烷酮进一发生羟醛缩合反应。
[0032]
通过加入甲醇作为溶剂先与1-苄基-3-吡咯烷酮液混合,加入活性镍,有利于1-苄基-3-吡咯烷酮液与活性镍混合均匀,避免了1-苄基-3-吡咯烷酮液反应时出现少量1-苄基-3-吡咯烷酮液活性较低导致产生1-苄基-3-吡咯烷醇;加入液氨加热后再加入氢气,既实现液氨与1-苄基-3-吡咯烷酮液混合均匀,同时省去了氢气前通惰性气体的过程,实现氨气与氢气混合相对均匀,有利于产应的快速反应,同时避免了局部氢气含量过高产生1-苄基-3-吡咯烷醇;从而进一步提高的1-苄基-3-氨基吡咯烷,另节约了加入惰性气体的成本;
[0033]
同时又避免了过量的氨气不容易收集,最终以甲醇氨的形式进行收集,节约了成本且环保。
[0034]
在通过逐步提高氢气压力,避免副反应的同时,在最后进行加热时,由于反应活性突然升高,在加氢反应时容易出现非定向加氢,容易出现副产品;
[0035]
因此在逐步提高氢气压力前通过将1-苄基-3-氨基吡咯烷盐化定向提高了1-苄基-3-氨基吡咯烷的反应活性,然后通过设定第四温度定向提高吡咯烷基的活性,实现了定向氢化,避免了副反应产生3-甲氨基吡咯烷,同时降低反应温度降低了反应成本。
[0036]
进一步的,第1)反应中所述第一温度为80-85℃,所述第一时长为4-4.5小时。
[0037]
采用上述进一步技术方案的有益效果在于,在此温度下有利于苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺的合成。
[0038]
进一步的,第2)反应中所述第二温度为20-30℃;所述第三温度为25-30℃,所述第二时长为3-4小时。
[0039]
采用上述进一步技术方案的有益效果在于,通过所述第二温度、第三温度有利于实现苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺在降低活性后,缓慢与乙醇钠反应,反应均匀,避免了在后续反应过程中副反应产生1-苄基-3-吡咯烷醇。
[0040]
进一步的,第2)反应中所述第一ph为2-3;所述第二ph为8-10,所述第三ph为8-9。
[0041]
采用上述进一步技术方案的有益效果在于,有利于在反应过程中避免副反应产生1-苄基-3-吡咯烷醇,同时抑制了得到的1-苄基-3-吡咯烷酮进一发生羟醛缩合反应。
[0042]
进一步的,第5)反应中所述第四温度为70-100℃。
[0043]
采用上述进一步技术方案的有益效果在于,第5)反应中在逐步提高氢气压力前通过将1-苄基-3-氨基吡咯烷盐化定向提高了1-苄基-3-氨基吡咯烷的反应活性,然后将反应时温度设定到70-100℃,定向提高吡咯烷基的活性,实现了定向氢化,避免了副反应产生3-甲氨基吡咯烷,同时降低反应温度降低了反应成本。
[0044]
进一步的,第3)反应中通入液氨具体过程为,分两步通入液氨,第一步通入所需添加液氨总量的35-45%的液氨,第二步通入液氨之前关闭反应容器上的排空阀。
[0045]
采用上述进一步技术方案的有益效果在于,通过分两步通入液氨,第一步添加35-45%的液氨,既有利于液氨与反应溶液混合均匀,同时实现了加入氢气前的对反应容器中空气排出;
[0046]
从而避免了在加入氢气前通入氮气存在的问题,具体为反应容器内反映溶液上方气体中氨气与氢气混合相对不均匀,且容易出现反应溶液局部与氢气直接单独接触,副反应产生1-苄基-3-吡咯烷醇;
[0047]
同时节约的氮气,降低了成本。
[0048]
进一步的,第3)反应中通入氢气具体过程为,分两步通入氢气,第一步通入氢气,反应容器内压力达到1.0-1.2mpa,然后开启搅拌;
[0049]
反应容器内温度为60-90℃、压力为1.5mpa-3.0mpa时,进行保温氢化反应;然后进行第二步通入氢气,提高压力为2.0-3.0mpa。
[0050]
采用上述进一步技术方案的有益效果在于,分两步通入氢气,第一步通入氢气,反应容器内压力达到1.0-1.2mpa,既满足了反应过程中氢气,且有利于不出现氢气局部过量问题,通过在加热过程中氨气的挥发,反应容器内压力达到1.0-1.2mpa,因此既实现了反映所需要的压力,同时避免因氢气局部过量出现副反用产生1-苄基-3-吡咯烷醇。
[0051]
进一步的,第1)反应中所述苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺的产率为88-90%;
[0052]
和/或
[0053]
第2)反应中所述1-苄基-3-吡咯烷酮的产率为92-99%;
[0054]
和/或
[0055]
第3)反应中所述1-苄基-3-氨基吡咯烷的产率为90-99%;
[0056]
和/或
[0057]
第5)3-氨基吡咯烷的产率为95-90%;
[0058]
所述苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺的产率为以丙烯酸乙酯的理论产出的苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺量为基准计算得到;
[0059]
所述1-苄基-3-吡咯烷酮的产率为以苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺的理论产出1-苄基-3-吡咯烷酮量为基准计算得到;
[0060]
所述1-苄基-3-氨基吡咯烷的产率为以1-苄基-3-吡咯烷酮的理论产出1-苄基-3-氨基吡咯烷量为基准计算得到;
[0061]
所述3-氨基吡咯烷的产率为以1-苄基-3-氨基吡咯烷的理论产出3-氨基吡咯烷量为基准计算得到。
[0062]
采用上述进一步技术方案的有益效果在于,所述制备方法可以有效实现上述产
率,明显提高了成品的产出比率,且降低了杂质的含量。
[0063]
进一步的,第4)反应中所述成盐反应具体过程为:
[0064]
将第3)反应中得到>50℃所述1-苄基-3-氨基吡咯烷液、异丙醇加入反应容器中;当反应容器内溶液降温到≤50℃时,然后向反应容器中加入氯化氢,并对反应溶液降温;
[0065]
当反应溶液ph小于6后,再加入第3)反应中得到>50℃所述1-苄基-3-氨基吡咯烷液,调节反应溶液ph为6-7;
[0066]
反应溶液降温小于30℃后,进行分离、干燥、重结晶得到1-苄基-3-氨基吡咯烷盐。
[0067]
采用上述进一步技术方案的有益效果在于,通过当反应溶液ph小于6后,再加入第3)反应中得到>50℃所述1-苄基-3-氨基吡咯烷液,调节反应溶液ph为6-7;既实现了1-苄基-3-氨基吡咯烷盐制备,同时降低了降温过程,节约了成本,缩短了工艺时间。
[0068]
进一步的,第6)反应中在进行蒸馏、干燥之后还包括3-氨基吡咯烷二盐酸盐精制,具体过程为:
[0069]
将甲醇、第6)反应中干燥之后还包括3-氨基吡咯烷二盐酸盐依次加入反应容器中,对反应溶液进行加热到64-65℃,保温58-62min;然后过滤;过滤后降温结晶,分两次降温,第一次降温到30-35℃,第二次降温到<5℃;结晶结束后进行干燥,即得。
[0070]
采用上述进一步技术方案的有益效果在于,实现3-氨基吡咯烷二盐酸盐的精制,提高了3-氨基吡咯烷二盐酸盐纯度。
具体实施方式
[0071]
为了更好的了解本发明的技术方案,下面结合具体实施例对本发明作进一步说明。
[0072]
实施例一:
[0073]
本实施例提供了一种3-氨基吡咯烷二盐酸盐合成方法,包括以下步骤:
[0074]
1)制备苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺液;
[0075]
(1)用真空泵依次抽入甲苯(可回收套用)、苄胺入反应罐;夹层通冷却水或冷盐水;
[0076]
(2)搅拌下分次抽入丙烯酸乙酯入反应罐中,加料完毕后,自然升温至平稳,继续搅拌10-15小时;
[0077]
(3)在40℃以内,充入氯化氢79.0kg,加料结束,继续搅拌反应4-6小时;
[0078]
(4)开启离心机,离心分离;滤饼用甲苯洗涤;所有滤液一并入反应罐;抽入氯乙酸乙酯、三乙胺;
[0079]
(5)抽料完毕,缓慢升温至80-85℃,保温4-4.5小时;
[0080]
(6)保温完毕,加适量的饮用水,搅拌10-20分钟,停止搅拌,静置2-2.5小时,放掉下层的水层,上层转移至蒸馏罐;
[0081]
(7)升温蒸出甲苯,160℃以内蒸馏至无液体流出后,继续减压蒸馏至不再有液体流出后,夹层通冷却水降温至30-40℃;
[0082]
(8)打开搅拌,抽入无水乙醇,搅拌10分钟,打开放料阀,放入周转桶中得苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺液,称量复核填物料卡;
[0083]
(9)结晶:将上述苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺液,转移至
不锈钢桶或周转桶,冷冻结晶48-96小时,冷冻结束将上层液体倒出并用少量冷冻无水乙醇洗涤后,晶体加热熔化,全部溶解后,抽入蒸馏罐120℃下减压蒸出无水乙醇,蒸馏结束,夹套通循环水降温至30-40℃得结晶后苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺液放入周转桶中,称重复核;
[0084]
物料添加质量比例:
[0085]
甲苯:苄胺:丙烯酸乙酯:氯化氢:氯乙酸乙酯:三乙胺为2:2:1:0.316:1.264:1.02;
[0086]
所述苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺的产率为89%;所述苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺的产率为以丙烯酸乙酯的理论产出的苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺量为基准计算得到;
[0087]
2)制备1-苄基-3-吡咯烷酮液;
[0088]
(1)用真空泵抽p1液、甲苯入反应罐中,夹层通冷却水,搅拌下加入乙醇钠,控制温度在20-30℃之间,加料完毕25-30℃保温反应3-4小时;
[0089]
(2)将上述澄清反应液抽入已加有纯化水、盐酸的反应罐内,抽料结束后,搅拌5-10分钟,测罐内的ph应为2-3,如果不是应加盐酸调至ph2-3,搅拌5-10分钟,静止1-1.5小时,将下层水溶液放入周转桶中或反应罐内暂存(上层溶液抽入另一反应罐进行蒸馏)。再将周转桶中水溶液抽入反应罐内,开动搅拌,升温蒸出甲苯水溶液,在98-100℃保温4.5小时,通冷却水降温到50-60℃;
[0090]
(3)用饱和氢氧化钠水溶液调ph8-10,用少量冰醋酸调ph至8-9,静置5-6小时,将下层放入化学污水管道,上层放入桶中得1-苄基-3-吡咯烷酮液,称重复核,填物料卡待验;
[0091]
物料添加质量比例:
[0092]
苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺:甲苯:乙醇钠:纯化水比例为1:1:0.28:1:1.5;
[0093]
所述1-苄基-3-吡咯烷酮的产率为96%;所述1-苄基-3-吡咯烷酮的产率为以苄基-(3-乙氧基-3-烯基)-(1-乙烯基乙氧基甲基)胺的理论产出1-苄基-3-吡咯烷酮量为基准计算得到;
[0094]
3)1-苄基-3-氨基吡咯烷液制备;
[0095]
投料过程:
[0096]
(1)确认蒸汽、冷却水、负压空气完好,罐体无积料,罐内无氮气压,关闭液氨进料针型阀、排空阀、氢气进气阀、氮气进气阀,打开真空泵、负压空气阀、进料阀,抽部分甲醇,(质量为100kg)入反应罐;
[0097]
(2)抽入1-苄基-3-吡咯烷酮液入反应罐;
[0098]
(3)抽入部分甲醇(质量为107kg)入反应罐,立即关闭“负压空气阀”;
[0099]
(4)活性镍的制备:
[0100]
向干燥的容器,加入第一氢氧化钠水溶液,所述第一氢氧化钠溶液中氢氧化钠固体质量:纯化水质量为1.3份:3份;加完后,向容器中缓慢加入镍铝合金质量为1份;静置分离,倒掉水层,再次加入第二氢氧化钠水溶液,所述第二氢氧化钠溶液中氢氧化钠固体质量:纯化水质量为1份:3份,搅拌、静置、分离,倒掉水层,用水洗涤至ph为8-9,用纯化水洗涤三次,用甲醇洗涤三次,装入有甲醇的容器;于阴凉干燥安全地方密封贮存备用;
[0101]
(4)加活性镍:确认进料管连接处绝对密封,将进料管插入装有甲醇的活性镍周转桶底部,打开负压空气阀,边搅边抽活性镍与剩余甲醇加入反应罐,抽活性镍过程中,保证活性镍周转桶内有足量甲醇,直至抽料结束,立即关闭“进料阀”;
[0102]
(5)关闭“负压空气阀”,打开“排空阀”;
[0103]
充液氨过程:
[0104]
(1)打开搅拌,10转/min,通冷却水降温,打开液氨罐阀门、液氨管路进料阀、液氨进料阀,充入液氨25kg后,关闭“排空阀”,充入剩余的液氨38kg,关闭液氨罐阀门、液氨进料阀门,关闭冷却水。
[0105]
(2)打开蒸汽缓慢升温5min,关闭出料口处的“高压球阀”;
[0106]
氢化过程:
[0107]
(1)打开氢气瓶阀门,调节罐体氢气阀门,使罐内压力达到1.0mpa,关闭罐体氢气阀门,设置搅拌转速为30转/min,同时升温,在60-90℃、1.5mpa-3.0mpa,保温氢化反应,最后提高氢压2.0-3.0mpa,当温度上升,压力同时上升,反应结束;
[0108]
(2)关闭氢气瓶阀门;
[0109]
(3)夹套通冷却水降温至20-30℃后,打开排空阀,卸掉余压;
[0110]
放料过程:
[0111]
(1)用氮气置换两次,最终保证罐内压力为0.3mpa-0.4mpa;
[0112]
(2)放料:打开过滤器进料阀——过滤器排空阀(排空阀压力<0.4mpa)——罐体出料高压阀、出料球阀,待过滤器排空阀出料后,关闭过滤器排空阀,打开过滤器出料阀将滤液转入周转桶;
[0113]
粗蒸过程:
[0114]
将氢化1-苄基-3-氨基吡咯烷液抽入反应罐,加热升温减压蒸出甲醇氨,150℃以下蒸馏至无馏份,降温70-80℃后装入周转桶内,送下一岗位;
[0115]
高温蒸馏过程:
[0116]
将粗蒸1-苄基-3-氨基吡咯烷液抽入高温蒸馏罐,减压加热升温至开始蒸出淡黄色液体,开始收集,至液体深棕红色为止,得高温蒸馏1-苄基-3-氨基吡咯烷液,放于周转桶中,称重复核,填好物料卡待验;
[0117]
物料添加质量比例:
[0118]
1-苄基-3-吡咯烷酮:液氨的比例为1:0.84;
[0119]
反应中所述1-苄基-3-氨基吡咯烷的产率为96%;所述1-苄基-3-氨基吡咯烷的产率为以1-苄基-3-吡咯烷酮的理论产出1-苄基-3-氨基吡咯烷量为基准计算得到;
[0120]
4)1-苄基-3-氨基吡咯烷盐的制备;
[0121]
投料过程:
[0122]
(1)抽高温蒸馏1-苄基-3-氨基吡咯烷液入反应罐,打开搅拌;
[0123]
(2)抽异丙醇入反应罐;
[0124]
(3)50℃以内充入氯化氢入反应罐,通冷盐水,自然反应,临近理论投料量,测反应液ph,使反应液ph达酸性;
[0125]
(4)抽少量高温蒸馏1-苄基-3-氨基吡咯烷液入反应罐,测反应液ph,使反应液ph达6-7;
[0126]
(6)继续降温至30℃以内,视物料粘稠度,可适当补加异丙醇(回收),准备分离;
[0127]
分离过程:
[0128]
离心机进料量由放料阀控制,离心甩滤至无液体流出,甩滤完毕,将固态离心物装入无毒聚乙烯袋中,称重复核送去干燥;
[0129]
干燥:
[0130]
将所得固体物料放入干燥箱内,40-50℃之间干燥8-10小时,干燥结束后,降温至20-30℃,用无毒聚乙烯袋包装后,称重复核挂填好物料卡,送入中间站;
[0131]
1-苄基-3-氨基吡咯烷盐母液提取:
[0132]
用真空泵将母液抽至反应罐,打开搅拌。抽异丙醇入反应罐。50℃以内充入氯化氢入反应罐,通冷盐水,自然反应,测反应液ph,使反应液ph达酸性。抽少量高温蒸馏1-苄基-3-氨基吡咯烷液入反应罐,测反应液ph,使反应液ph达6-7。继续降温至30℃以内,视物料粘稠度,可适当补加异丙醇,准备分离;
[0133]
分离过程:
[0134]
离心机进料量由放料阀控制,离心甩滤至无液体流出,甩滤完毕,将固态离心物装入无毒聚乙烯袋中,称重复核送去干燥;
[0135]
干燥:将所得固体物料放入干燥箱内,40-50℃之间干燥8-10小时,干燥结束后,降温至20-30℃,得到1-苄基-3-氨基吡咯烷,用无毒聚乙烯袋包装后,称重复核挂填好物料卡,送入中间站;
[0136]
5)3-氨基吡咯烷液的制备:
[0137]
(1)抽甲醇至高压反应罐内,搅拌下加入1-苄基-3-氨基吡咯烷盐;
[0138]
(2)将打湿的钯炭(钯炭1kg,纯化水10kg)抽入至高压反应罐内;
[0139]
(3)抽入剩余的纯化水至高压反应罐内;
[0140]
(4)开启搅拌,用氮气置换2次,每次0.3-0.5mpa,搅拌3-5分钟,用氢气置换2次,每次0.3-0.5mpa,搅拌3-5分钟,然后提高氢压至2.0-3.0mpa,加热升温至吸氢开始记录反应温度,压力下降,补充氢压,在70-100℃,1.5mpa-3.0mpa,保温氢化反应,最后提高氢压2.0-3.0mpa,当温度上升,压力同时上升,反应结束,夹层通冷却水降温至20-30℃,泄掉氢压;
[0141]
(5)放料:用氮气置换两次,最终保证罐内压力为0.3mpa;打开过滤器进料阀——过滤器排空阀(排空阀压力<0.3mpa)——罐体出料高压阀、出料球阀,待过滤器排空阀出料后,关闭过滤器排空阀,打开过滤器出料阀将滤液转入周转桶;
[0142]
反应中所述3-氨基吡咯烷的产率为98%;所述3-氨基吡咯烷的产率为以1-苄基-3-氨基吡咯烷的理论产出3-氨基吡咯烷量为基准计算得到;
[0143]
6)3-氨基吡咯烷二盐酸盐的制备;
[0144]
3-氨基吡咯烷二盐酸盐粗品的制备:
[0145]
(1)反应过程:将p4液抽至反应罐中,夹层通冷却水,用盐酸调ph至2-3后,封罐升温,130℃以下常压蒸馏至馏份不出,减压蒸馏份至不出。再抽入热甲苯,130℃以下常压蒸馏至不出,减压蒸馏至不出,用热无水乙醇分散,降温至40℃以下;
[0146]
(2)分离过程:离心机进料量由放料阀控制,离心甩滤至无液体流出,用适量的无水乙醇洗涤,离心甩滤至无液体流出,甩滤完毕,将固态离心物装入无毒聚乙烯袋中,称重复核送去干燥;
[0147]
(3)干燥:将所得固体物料放入干燥箱内,40-50℃之间干燥1-2小时,干燥结束后,降温至20-30℃,用无毒聚乙烯袋包装后,称重复核挂填好物料卡,送入中间站;
[0148]
3-氨基吡咯烷二盐酸盐精制:
[0149]
将甲醇抽入反应罐中,开动搅拌,缓慢加入3-氨基吡咯烷二盐酸盐粗品,封罐升温加热至澄清,在64-65℃,保温1小时,热过滤至另一反应罐,通冷却水降温至30-35℃,通冷盐水降温至5℃以下,结晶4小时,结晶结束,开启离心机,分离,所得白色或类白色结晶装入聚乙烯袋中,称重复核,填好物料卡,送干燥;
[0150]
干燥:将所得固体物料放入干燥箱内,40-50℃之间干燥2-4小时,干燥结束后,降温至20-30℃,用聚乙烯袋包装后,称重复核挂填好物料卡,送检。
[0151]
以上描述仅为本技术的较佳实施例以及对所运用技术原理的说明。本领域技术人员应当理解,本技术中所涉及的发明范围,并不限于上述技术特征的特定组合而成的技术方案,同时也应涵盖在不脱离所述发明构思的情况下,由上述技术特征或其等同特征进行任意组合而形成的其它技术方案。例如上述特征与本技术中公开的(但不限于)具有类似功能。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1