一种非对称氧化磷吡啶三嗪类衍生物及其合成方法
1.本发明涉及有机合成技术领域,具体涉及一种非对称氧化磷吡啶三嗪类衍生物及其合成方法。
背景技术:
2.核燃料的有效处理、处置是确保核能可持续发展的关键因素,特别是其中具有长期强放射性锕系元素的分离问题。目前国际上采用的普雷克斯流程(purex,plutonium uranium reduction extraction),可将乏燃料中大部分的铀和钚(约占乏燃料总量的95%)分离后循环利用。而铀钚分离后所产生的废液其中硝酸浓度为3-4m也就是所谓的高放废液(high active liquid waste,hlw),其中仍含长寿命次锕系元素(如am(iii)、cm(iii))却是高放废液的长期放射性和辐射毒害的主要来源。因此非常有必要将长寿命的三价次锕系元素进行分离-嬗变(partitioning&transmutation,p&t)从而消除三价次锕系元素的长期放射性污染的问题。但直接将锕系元素和镧系元素从高放废液中进行高效分离难度非常大,到目前为止也尚无工业化的镧锕分离流程。
3.为了降低三价镧系和锕系从高放废液中进行分离的难度,人们采取两步法来实现分离嬗变的目标。首先第一步采用含有o的配体将三价的次锕系元素am(iii)/cm(iii)以及三价的ln(iii)离子进行共同萃取同高放废液中其他裂片元素进行分离,然后再采用含有n或者s的配体实现三价的am(iii)/cm(iii)以及ln(iii)之间的相互分离,分离流程较为繁琐,仍在存在萃取效率低、化学稳定性低、产生二次废物多等缺点仍需要进一步的改进,所采用的的配体合成步骤复杂,且成本较高。
4.cn109750164a公开了一种用于乏燃料后处理中三价锕系与镧系选择性萃取分离方法,该方法以ntaamide(n-oct)作为萃取剂,煤油为稀释剂,水溶性配体tee-bisdga为洗涤剂,以羟基络合剂或氨羧络合剂或硝酸溶液为范翠及,利用多级萃取工艺实现镧系和锕系元素的选择性萃取分离。但其萃取过程复杂,需要萃取级数10-15次,洗涤段的级数为3~6级,反萃段的级数为4-8级,耗费设备和时间。
5.发明人在前期研究过程中(cn112458283a)发现邻菲罗啉氧化磷对三价锕系和三价稀土离子具有较好的共分离萃取效果。但邻菲罗啉氧化磷无法将三价次锕系离子和镧系离子相互分开。因此从乏燃料中实现三价次锕系元素与镧系分离的难题仍然需要进一步探索。
技术实现要素:
6.本发明针对乏燃料中分离三价次锕系元素与镧系的难题,合成了一种同时具有烷基氧化磷和吡啶三嗪环结构,耐强酸水解,具有萃取能力强、萃取速率快等优点,且该衍生物合成步骤简单,在核工业废液特别是高放废液中分离三价镧系和锕系共萃取分离领域中具有良好的应用前景。
7.为实现上述目的,本发明采用的技术方案是:
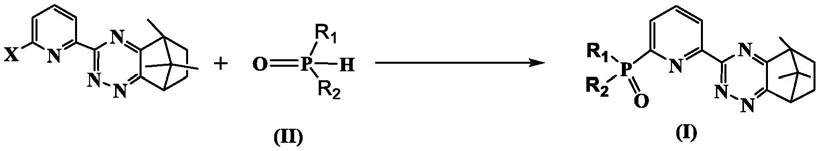
8.一种非对称氧化磷吡啶三嗪类衍生物,具有如式i所示的结构式:
[0009][0010]
其中,r1为苯基或取代苯基;r2为苯基、c1-c10的直链烷基或支链烷基。
[0011]
本发明中设计的萃取剂在结构上兼有氧化磷和三嗪环功能基团,使得该萃取剂具有两种官能团的功能特性。现有技术中曾有报道只含有氧化磷官能团的具有对称结构的二苯基氧化磷吡嗪萃取剂,但其对am(iii)的萃取能力较弱(d
am
《1),且当溶液中的酸度大于1.0m时该配体丧失了对am(iii)的萃取能力(d
am
《0.1)。在本发明中再含有氧化磷结构的萃取剂上引入了三嗪环的结构,可显著增强其在高硝酸浓度下对am(iii)的萃取能力。同时还避免了只含有三嗪环的对称性配体其在酸度大于1.0m的条件下萃取am(iii)时容易发生质子化出现沉淀,丧失萃取能力的问题。本发明中的非对称萃取剂充分利用氧化磷和三嗪环的优点克服其各自的不足,相互协同,从而获得针对三价镧系或锕系离子均具有优异萃取分离效果的萃取剂。
[0012]
优选地,r1为苯基或取代苯基;r2为苯基、乙基、正丁基、正辛基或叔丁基。
[0013]
进一步优选地,r1为苯基,r2为苯基或正丁基。此两种取代基的配体合成产率较高,具有成本优势,且萃取效果也较为优异。
[0014]
本发明还提供所述的非对称氧化磷吡啶三嗪类衍生物的合成方法,包括步骤:
[0015]
步骤1,以6-卤代-2-氰基吡啶与一水合肼反应,得到中间产物a;
[0016][0017]
其中x表示卤素,包括br或cl或i;
[0018]
步骤2,向樟脑醌的溶液中滴加中间产物a,反应后得滤液旋干、纯化,得到中间产物b;反应式如下:
[0019][0020]
步骤3,中间产物b与式ii所示的氧化磷在溶液中溶解后,在催化剂和碳酸铯的作用下回流反应,产物洗涤、纯化得到所述非对称氧化磷吡啶三嗪类衍生物;
[0021][0022]
步骤3的反应式如下:
[0023][0024]
步骤1中,反应溶剂包括水和/或乙醇,反应温度为5-40℃,反应时间为30min-2h。
[0025]
步骤1中,一水合肼与卤代-2-氰基吡啶的摩尔比为(1.5~5):1;
[0026]
所述卤代-2-氰基吡啶包括6-溴-2-氰基吡啶、6-氯-2-氰基吡啶、6-碘-2-氰基吡啶中任一种。
[0027]
步骤2中,樟脑醌与中间产物a摩尔比为(1.2~2.0):1;
[0028]
步骤2中反应的溶剂包括乙醇和/或四氢呋喃;
[0029]
优选地,步骤2反应在高纯惰性气体保护下进行;tlc(薄层色谱分析)跟踪反应至原料反应完全。高纯惰性气体包括氮气或氩气。
[0030]
步骤2中纯化过程采用硅胶柱色谱分离法进行纯化,淋洗剂包括乙酸乙酯、石油醚、二氯甲烷、甲醇中一种或多种的混合物。
[0031]
优选地,淋洗剂为乙酸乙酯、石油醚和二氯甲烷的混合物或二氯甲烷、乙酸乙酯和甲醇的混合物,可通过薄层色谱分析其分离效果进行淋洗剂中各溶剂的比例确认。
[0032]
步骤3中,中间产物b与所述氧化磷的摩尔比1:(1.2~2.5),为了提高中间产物b的转化效率,使氧化磷进行过量反应。
[0033]
所述催化剂包括1,1'-双(二苯基膦)二茂铁(dppf)和醋酸钯或dppf和新戊酸钯,催化剂的摩尔量为反应原料的1-3%;碳酸铯的添加质量为反应原料质量的2-5倍。
[0034]
与现有技术相比,本发明具有以下有益效果:
[0035]
本发明中的非对称氧化磷吡啶三嗪类衍生物结构性能稳定,针对三价镧系和锕系元素具有优异的萃取效果,萃取能力强且快;同时该衍生物合成步骤简单,在核工业废液特别是高放废液中分离三价镧系和锕系共萃取分离领域中具有良好的应用前景。
附图说明
[0036]
图1为实施例1中ph2ca-tppo的核磁谱图。
[0037]
图2为实施例1中ph2ca-tppo的质谱图。
[0038]
图3为实施例1中ph2ca-tppo的紫外光谱图。
[0039]
图4为实施例2中buphca-tppo的核磁谱图。
[0040]
图5为实施例2中buphca-tppo的质谱图。
[0041]
图6为应用例1中ph2ca-tppo在不同硝酸浓度对am(iii)和eu(iii)的萃取效果。
具体实施方式
[0042]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。本领域技术人员在理解本发明的技术方案基础上进行修改或等同替换,而未脱离本发明技术方案的精神和范围,均应涵盖在本发明的保护范围内。
[0043]
以下具体实施方式中所采用的原料均购于市场,未经处理直接使用。
[0044]
实施例1 ph2ca-tppo的合成
[0045]
步骤1,将2.5g的6-溴-2-氰基吡啶与一水合肼10.0ml置于反应瓶中,并加入10.0ml去离子水中于25℃条件下反应1.0h。反应后直接过滤得到固体粉末,再用去离子水洗涤三次,然后于60℃下干燥过夜得到中间产物2,约为2.0g。
[0046][0047]
步骤2,将2.3g的樟脑醌溶于超干乙醇或四氢呋喃、并通过恒压漏斗滴加至含有2.0g溴代单吡啶胺腙(步骤1制备的中间产物2)的超干乙醇悬浮溶液,,滴加完成后反应。先在高纯氩气保护条件下回流反应3h,然后在常温条件下搅拌过夜,tlc跟踪反应直至原料反应完全。
[0048]
反应后得到红棕色溶液经沙星漏斗过滤除去固体杂质,并将滤液旋干得到油状产物,加入300-400目硅胶8g制备干法柱色谱样品,采用乙酸乙酯/石油醚/二氯甲烷(体积比1/7/2)作为淋洗剂,通过硅胶柱色谱分离技术进行分离纯化,得到中间产物4,旋干溶剂得到纯品1.2g,产率30%。
[0049][0050]
步骤3,将1.0g中间产物与0.48g二苯基氧化磷溶解于50.0ml超干甲苯溶液中并向溶液中加入醋酸钯28.0mg(0.122mmol),dppf 160.0mg(0.244mmol),碳酸铯3.0g在氩气的保护下,抽真空处理,在110℃下回流反应16h。反应完成后经抽滤、旋转蒸发仪除去溶剂甲苯。在粗产品中加入二氯甲烷和饱和nacl溶液,洗涤萃取多次后合并有机相,得到棕色油状物,采用二氯甲烷/乙酸乙酯/甲醇(v/v/v=30:30:1)作为淋洗剂经过柱色谱分离,得到产品ph2ca-tppo棕色油状物0.5g。
[0051][0052]
(1)ph2ca-tppo的1h nmr表征
[0053]
对ph2ca-tppo进行核磁表征,结果如图1所示,其中各峰归属为:1hnmr(500mhz,cd3od,298k):
[0054]
δ(ppm):8.61(m,1h,arh),8.34(m,1h,arh),8.21(m,1h,arh),8.04(m,4h,arh),7.59(m,2h,arh),7.52(m,4h,arh),3.25(d,1h,ch),2.42(m,1h,ch),2.18(m,1h,ch),1.44(s,3h,ch3),1.39(d,2h,ch2),1.17(s,3h,ch3),0.65(s,3h,ch3);1h nmr结果表明表明该化合物含有27个氢原子,与其理论值相符合,说明成功合成了化合物ph2ca-tppo。
[0055]
(2)ph2ca-tppo的esi-ms表征
[0056]
对ph2ca-tppo进行质谱表征,结果如图2所示,467.23为化合物结合一个氢离子的
分子离子峰[m+h]
+
,该结果表明其与该化合物ph2ca-tppo的分子量理论值466.5相吻合。
[0057]
(3)ph2ca-tppo的紫外-可见光谱表征
[0058]
对ph2ca-tppo紫外-可见光谱表征,结果如图3所示,ph2ca-tppo在251.2nm和266.6nm处分别有一个最大吸收峰,该结果表明其与该化合物结构中含有芳香基团。
[0059]
实施例2 buphca-tppo的合成
[0060]
将实施例1步骤2制备得到的中间产物4取1g,与0.8g丁基苯基氧化磷溶解到50.0ml超干甲苯溶液中并向溶液中加入醋酸钯28.0mg(0.122mmol),dppf 1,1'-双(二苯基膦)二茂铁160.0mg(0.244mmol),碳酸铯3.0g在氩气的保护下,抽真空处理,在110℃下回流反应24h。反应完成后经抽滤、旋转蒸发仪除去溶剂甲苯。在粗产品中加入二氯甲烷和饱和nacl溶液,洗涤萃取多次后合并有机相,得到棕色油状物,采用二氯甲烷/乙酸乙酯/甲醇(v/v/v=30:30:1)作为淋洗剂经过柱色谱分离,得到产品buphca-tppo棕色油状物0.8g。
[0061][0062]
(1)buphca-tppo的1h nmr表征
[0063]
对buphca-tppo进行核磁表征,结果如图4所示,其中各峰归属为:1h nmr(500mhz,cd3od,298k):
[0064]
δ(ppm):8.62(m,1h,arh),8.17(m,2h,arh),8.07(m,1h,arh),7.55(m,3h,arh),3.30(d,1h,ch),2.60-2.70(m,2h,ch2),2.20(m,1h,ch),2.01(m,1h,ch),1.55(m,2h,ch2),1.46-1.31(m,7h,2
×
ch2,1
×
ch3),1.18(s,3h,1
×
ch3),0.99(d,3h,ch3),0.90(m,3h,ch3),0.68(s,3h,ch3);1h nmr结果表明该化合物为非对称结构6个甲基形成6个峰,表明该化合物含有31个氢原子,与其理论值相符合,说明成功合成了化合物buphca-tppo。
[0065]
(2)buphca-tppo的esi-ms表征
[0066]
对buphca-tppo进行质谱表征,结果如图5所示,447.21为化合物结合一个氢离子的分子离子峰[m+h]
+
,该结果表明其与该化合物buphca-tppo的分子量理论值446.5相吻合。
[0067]
应用例
[0068]
将实施例1制备的ph2ca-tppo溶解于3-硝基三氟甲苯溶剂中制备0.010摩尔/升的配体溶液作为萃取实验的有机相;水相则为含有放射性元素
241
am(iii)和
152,154
eu(iii)的硝酸水溶液,通过加入一定量的浓硝酸并采用去离子水稀释进行酸碱标定,配制成具有一系列硝酸浓度的含有金属离子的水溶液作为萃取的水相,其硝酸设定为0.1,0.5,1.0,2.0,3.0,4.0摩尔/升。
[0069]
萃取实验时,先分别取0.6ml含有
241
am(iii)和
152,154
eu(iii)水相的不同浓度的硝酸水溶液作为萃取过程中的水相。充分混合均匀后分别取出100μl含有
241
am(iii)和
152,154
eu(iii)的水相溶液置于液闪瓶中,采用液闪仪测量表萃取前水相中的
241
am(iii)和
152,154
eu(iii)的放射性活度。然后将剩下的含有
241
am(iii)和
152,154
eu(iii)的0.5ml的不
同浓度的硝酸水溶液分别与等体积的浓度为0.01m的ph2ca-tppo配体溶液的有机相进行混合,然后于室温下进行磁力搅拌混合,萃取达到平衡状态后对混合液进行离心分相,分相后对萃取后水相中
241
am和
152,154
eu(iii)的放射剂活度进行分析,分别得到0.1,0.5,1.0,2.0,3.0,4.0摩尔/升的硝酸浓度条件下,
241
am(iii)和
152,154
eu(iii)的萃取分配比d,如表1和图6所示。其中d
am(iii)
表示am(iii)的分配比,d
eu(iii)
表示eu(iii)的分配比,sf
am(iii)/eu(iii)
表示am(iii)相对于eu(iii)的分离系数。
[0070]
表1 ph2ca-tppo对
241
am(iii)和
152,154
eu(iii)的萃取效果
[0071][0072]
hno3浓度影响萃取分离结果
[0073]
ph2ca-tppo萃取结果如表1和图6所示,随着硝酸浓度的增大,ph2ca-tppo对
241
am和
152,154
eu(iii)的萃取分配比d逐渐增大,当硝酸浓度为3.0-4.0摩尔/升时,
241
am的萃取分配比达到最大约为10,
241
am的萃取率约为90%,该酸度与高放废液的酸度相吻合,说明该配体非常适合用于从高放废液中萃取分离三价次锕系元素。与此同时当酸度大于1.0m时,
241
am和
152,154
eu(iii)的萃取分离系数sf
am/eu
均高于15。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1