1,3,5-三取代-吡唑-4羧酸衍生物及其制备方法和应用
1.本发明涉及一种1,3,5-三取代-吡唑-4羧酸衍生物及其制备方法和应用,属于有机化学合成方法学领域。
背景技术:
2.多取代吡唑及其衍生物被认为是一种重要的药物支架,具有几乎所有类型的药理活性。这一基本药物骨架广泛存在于不同的治疗类药物中,如抗炎药塞来昔布、抗精神病药cdppb、抗焦虑药扎来普隆、抗肥胖药利莫那班以及cox-2选择性非甾体抗炎药(nsaid s)替泊沙林等。这些均表明了吡唑骨架具有很大的药物潜力。在过去的十年中,许多科学家和研究人员报道了一系列具有良好抗癌活性的吡唑衍生物,表明吡唑基序是开发新型抗癌药物的有力工具。
3.通常合成吡唑核的常用方法有:(a)肼及其衍生物在羰基体系上的环缩合反应,但此方法往往会生成一对区域异构体的混合物,且难以分离,增大目标产物提取成本;(b)由其他杂环体系如吡喃酮、呋喃二酮、嘧啶、嘧啶酮等来制备,然而,这些特殊的基底源限制了该方法的应用;(c)1,3-偶极环加成反应,与前两种方法相比,此种方法具有反应条件温和、操作过程简便、后处理步骤简单、产率高等特点,是合成吡唑类化合物最有前景的方法之一。
4.但现有的吡唑环类化合物仍无法满足人们日益增长的需要。
技术实现要素:
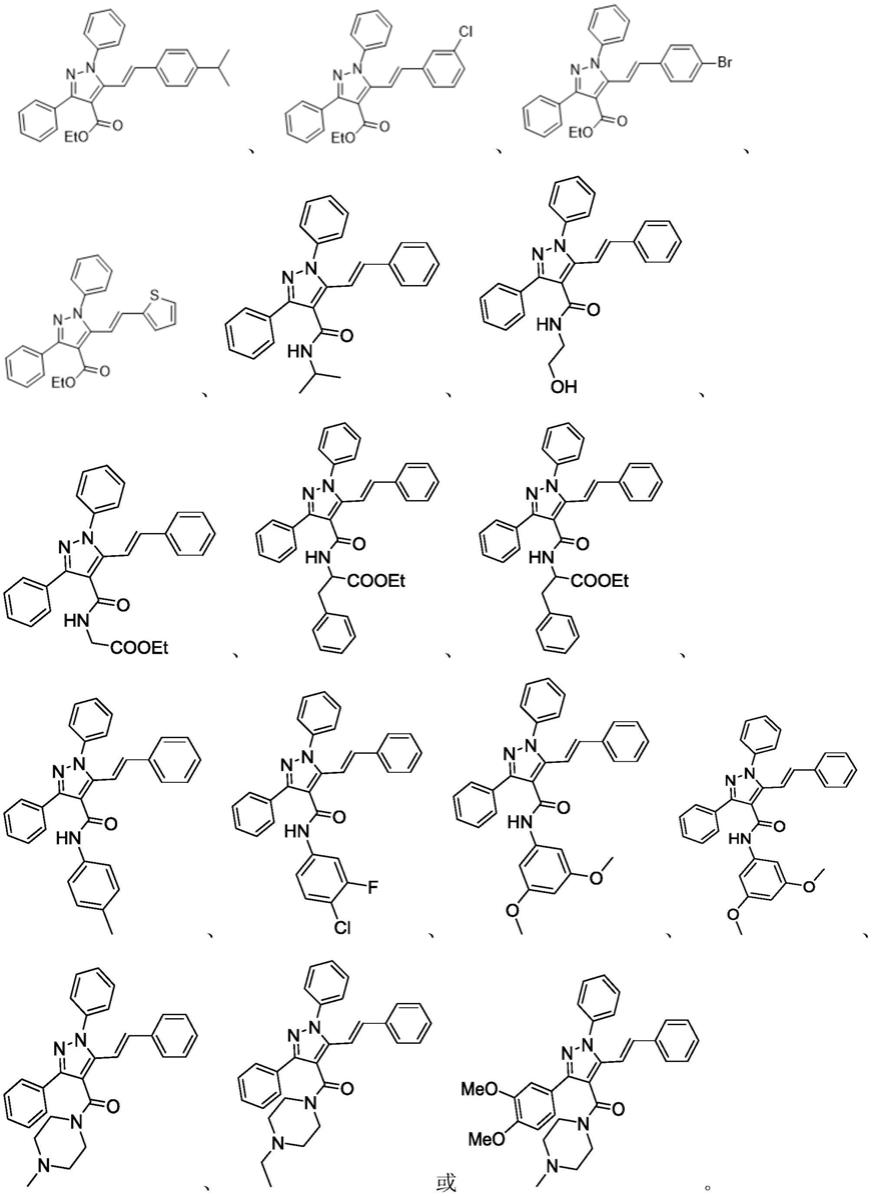
5.为进一步扩大吡唑环类化合物的dna编码化合物库(del)筛选的化学空间,本技术提出了一种1,3,5-三取代-吡唑-4-羧酸衍生物,其具有如下通式:
[0006][0007]
其中:r1为苯基、取代苯基或杂环基,r2为eto基(乙氧基)、氨基、苯胺基、取代胺苯基或杂环胺基,r3为苯基、取代苯基或杂环基,r4为苯基、取代苯基或杂环基。其中的杂环基优选吗啉基或哌嗪基。
[0008]
具体地,1,3,5-三取代-吡唑-4-羧酸衍生物为结构式为如下任意一种的化合物:
[0009]
[0010][0011]
多取代吡唑及其衍生物被认为是一种重要的药物支架,具有几乎所有类型的药理活性。这一基本药物骨架广泛存在于不同的治疗类药物中,如抗炎药塞来昔布、抗精神病药cdppb、抗焦虑药扎来普隆、抗肥胖药利莫那班以及cox-2选择性非甾体抗炎药(nsaid s)替泊沙林等。这些均表明了吡唑骨架具有很大的药物潜力。上述羧酸衍生物扩大了多取代吡唑衍生物的品种,为相应药物的选择提供了更大的选择范围。
[0012]
其次,本技术还公开了上述1,3,5-三取代-吡唑-4-羧酸衍生物的制备方法,其包括合成方法a和合成方法b;
[0013]
当r1为苯基、取代苯基或杂环基,r2为eto基,r4为苯基、取代苯基或杂环基时,1,3,5-三取代-吡唑-4-羧酸衍生物称为化合物a;当r1为苯基、取代苯基或杂环基,r2为氨基、苯胺基、取代苯胺基或杂环胺基,r4为苯基、取代苯基或杂环基时,1,3,5-三取代-吡唑-4-羧酸衍生物称为化合物b;
[0014]
其中合成方法a用于合成化合物a,合成方法b用于合成化合物b
[0015]
该合成方法a为:
[0016]
以联烯酸酯和n-苯基苯并腙酰氯为原料,以碳酸钾为催化剂,以二氯甲烷为溶剂进行反应所制得;具体的反应式如式(ⅰ):
[0017][0018]
在该合成化合物a中,r1优选为苯基、氟代苯基、氯代苯基、溴代苯基或杂环基。
[0019]
该合成方法b为:
[0020]
以联烯酸酯和n-苯基苯并腙酰氯为原料,以碳酸钾为催化剂,以二氯甲烷为溶剂进行反应,获得化合物a;
[0021]
再以化合物a和氢氧化锂为原料,四氢呋喃水溶液为溶剂,得到水解产物a;
[0022]
然后将水解产物a和亚硫酰氯溶解在chcl3中,进行反应并浓缩;将浓缩液溶解在二氯甲烷中,并滴加三乙胺和胺类化合物的混合溶液进行反应后,得到化合物b;
[0023]
其中的胺类化合物为2-丙胺、乙醇胺、甘氨酸乙酯盐酸盐、苯丙氨酸乙酯盐酸盐、苯胺、4-甲基苯胺、4-氯-3-氟苯胺、3,5-二甲氧基苯胺、吗啉、1-甲基哌嗪或1-乙基哌嗪。
[0024]
化合物a制备化合物b的反应式如式(ⅱ):
[0025]
[0026]
联烯酸酯是具有丙二烯结构的一类化合物,其具有充分的反应活性,本技术利用4-乙酰氧基联烯酸酯与n-苯基苯并腙酰氯的1,3-偶极环加成反应合成了一系列1,3,5-三取代吡唑-4-羧基酸衍生物。在此反应中,联烯酸酯c4位置的乙酰氧基可以通过消除一个hoac分子来促进环加成反应,使得反应在温和的条件下进行,且产率较高。且通过在4-乙酰氧基联烯酸酯上引入苯环及取代苯环,在上述吡唑环化合物的5位引入了不同取代的苯乙烯基,进一步扩大了吡唑环类化合物的dna编码化合物库(del)筛选的化学空间。
[0027]
因而在本发明中,我们以4-乙酰氧基联烯酸酯为亲偶极子,以n-苯基苯并腙酰氯为1,3-偶极子前驱体,通过简单的1,3-偶极环加成反应制备了一系列1,3,5-三取代-吡唑-4羧酸衍生物。
[0028]
具体地,为顺利地完成反应,其中该合成方法a具体包括如下步骤:
[0029]
(1.1)将n-苯基苯并腙酰氯溶于二氯甲烷中,然后加入碳酸钾,再逐滴加入联烯酸酯,在氩气保护下,置于室温下反应6-10小时;
[0030]
(1.2)反应完成后,向反应液中加入去离子水,用乙酸乙酯萃取后,合并有机相,再使用去离子水洗涤;收集有机相,然后用无水硫酸钠对有机相进行脱水,获得脱水有机相;
[0031]
(1.3)在脱水有机相中加入硅胶,旋干溶剂制沙,获得第一旋干物,随后以石油醚和乙酸乙酯的混合溶剂作为洗脱剂对第一旋干物进行柱层析梯度洗脱,收集检测到的所有产物的洗脱液,旋蒸脱除洗脱液中的混合溶剂后得到化合物a。
[0032]
合成方法b包括如下步骤:
[0033]
(2.1)将n-苯基苯并腙酰氯溶于二氯甲烷中,然后加入碳酸钾,再逐滴加入联烯酸酯,在氩气保护下,置于室温下反应6-10小时;
[0034]
(2.2)反应完成后,向反应液中加入去离子水,用乙酸乙酯萃取后,合并有机相,再使用去离子水洗涤3-5次;收集有机相,然后用无水硫酸钠对有机相进行脱水;获得脱水有机相;
[0035]
(2.3)在脱水有机相中加入硅胶,旋干溶剂制沙,获得第一旋干物,随后以石油醚和乙酸乙酯的混合溶剂作为洗脱剂对第一旋干物进行柱层析梯度洗脱,收集检测到的所有产物的洗脱液,旋蒸脱除洗脱液中的混合溶剂后得到化合物a;
[0036]
(2.4)将1,3,5-三取代-吡唑-4-羧酸衍生物和氢氧化锂溶于四氢呋喃水溶液中,得到衍生物混合液,将衍生物混合液油浴锅中110℃加热回流,进行水解反应,用薄层色谱法监测反应,直至1,3,5-三取代-吡唑-4-羧酸衍生物全部反应;水解反应的时间优选5小时;
[0037]
反应完成后,将四氢呋喃真空旋干,获得第二旋干物,在第二旋干物中加入稀盐酸调节ph至2,析出黄色固体,然后通过真空过滤收集产物并干燥以获得水解产物a;
[0038]
(2.5)将水解产物a和亚硫酰氯溶解在chcl3中,然后在80℃回流,当水解产物a被消耗完毕时,将混合物浓缩,得相应的酰氯化合物b;水解产物a和亚硫酰氯的摩尔比优选1:4;80℃回流时间优选4小时;
[0039]
将酰氯化合物b溶解在二氯甲烷中,并向二氯甲烷中滴加三乙胺和胺类化合物的混合溶液,保持搅拌,在0℃及氩气保护下进行胺化反应,反应结束后,在反应混合物中加入硅胶,旋干溶剂制沙,获得第三旋干物,随后通过pe:ea=5:1-1:1的柱色谱法对第三旋干物进行纯化,得到化合物b;胺化反应的时间优选为5小时。
[0040]
具体地,步骤(1.1)和(2.1)中,反应时间均为6-10小时。
[0041]
具体地,步骤(1.3)和(2.3)中,在石油醚和乙酸乙酯的混合溶剂中,石油醚与乙酸乙酯体积比均为5:1;n-苯基苯并腙酰氯、联烯酸酯以及碳酸钾三者的摩尔比均为1:1:1.2。
[0042]
具体地,步骤(2.4)中,1,3,5-三取代-吡唑-4-羧酸衍生物与氢氧化锂的摩尔比为1:4;四氢呋喃水溶液中,四氢呋喃与水的体积比为1:1;稀盐酸的浓度3mol/l。
[0043]
进一步,步骤(2.5)中,水解产物a与亚硫酰氯的摩尔比为1:4,酰氯化合物b与胺类化合物的摩尔比为1:1;三乙胺的浓度为0.5mmol/ml。
[0044]
再次,本技术还公开了所述1,3,5-三取代-吡唑-4-羧酸衍生物在细胞活性分析中的应用。该应用具体为:为便于描述,将1,3,5-三取代-吡唑-4-羧酸衍生物简称为羧酸衍生物:
[0045]
(1)接种细胞:用含10%胎牛血清的培养基制备单细胞悬液,每孔接种1000个du145细胞至96孔板中,每孔体积100ul;
[0046]
(2)配药:将羧酸衍生物(1mg,2.5μmol)置于1mldmso中溶解,配制成2500μm的第一羧酸衍生物溶液。随后在第一羧酸衍生物溶液中加入9ml ldmso将其稀释10倍,配置成250μm的第二羧酸衍生物溶液。接着再取1ml稀释后的250μm的第二羧酸衍生物溶液,加入4mldmso将其稀释5倍,配制成50μm的第三羧酸衍生物溶液。取2ml 50μm的第三羧酸衍生物溶液,加入3ml dms0将其稀释2.5倍,配制成20μm的第四羧酸衍生物溶液。取1m 20μm的第四羧酸衍生物溶液,加入1ml dms0将其稀释2倍,配制成10μm的第五羧酸衍生物溶液;
[0047]
(3)给药:细胞接种后24小时,每孔分别加入100ul第五羧酸衍生物溶液、第四羧酸衍生物溶液和第三羧酸衍生物溶液,孵育4天;
[0048]
(4)着色:培养4天后,每孔加入10ul mtt溶液(5mg/ml in pbs,ph=7.4);继续孵育4h终止培养,吸弃孔内培养上清,将悬浮细胞离心后吸弃孔内培养上清;每孔加入100ul dmso,摇晃,使晶体完全溶解;
[0049]
(5)比色法:选择570nm波长,在酶联免疫吸附仪上测量各孔的吸光值,记录结果,用graphpad软件处理数据,列入下表1,得到ic
50
。
[0050]
表1
[0051][0052]
有益效果
[0053]
本发明是全新的化合物,同时本发明提供该化合物的合成方法,该反应起始原料易得,条件温和,合成路线简短、操作方便,成本较低。该反应具有较好的化学选择性,产率高,并且可将该反应进行规模放大生产,因此在有机合成中具有较高的实用性。通过此法合成的1,3,5-三取代-吡唑-4羧酸衍生物具有潜在的生物活性,酯基水解酸化为相应的羧酸且羟基可以进行多种衍生化,在新药研发领域有较好的应用前景。
具体实施方式
[0054]
下面结合具体实施例来进一步描述本发明。
[0055]
实施例1
[0056]
(e)-1,3-二苯基-5-苯乙烯基-1h-吡唑-4-羧酸乙酯(iii-1),具体结构式如下:
[0057][0058]
合成方法具体如下:
[0059]
在25ml圆底烧瓶中加入n-苯基苯并腙酰氯(23mg,0.1mmol)、碳酸钾(17mg,0.12mmol)和3mlch2cl2,。随后在该混合液中滴加5-乙酰氧基-5-苯基五-2,3-二烯酸乙酯的ch2cl2溶液,在氩气保护下,并在室温下搅拌反应8h。
[0060]
反应完成后,向反应液中加入50ml去离子水,使用乙酸乙酯萃取后,合并有机相,再使用去离子水溶液洗涤3次;收集有机相,然后用无水硫酸钠对有机相进行脱水,获得脱
水有机相;
[0061]
在有机相中加入硅胶,旋干溶剂制沙,获得第一旋干物,再以石油醚:乙酸乙酯体积比为5:1的混合溶剂作为洗脱剂对第一旋干物进行柱层析洗脱,收集检测到的所有产物的洗脱液,旋蒸脱除洗脱液中的混合溶剂后得到黄色油状物的iii-1号27mg,收率70%。
[0062]
对本实施例中的iii-1号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0063]1h nmr(300mhz,chloroform-d)δ7.76(dd,j=8.0,1.6hz,2h),7.55
–
7.31(m,13h),7.30
–
7.26(m,1h),6.88(d,j=16.7hz,1h),4.28(q,j=7.1hz,2h),1.18(t,j=7.1hz,3h)。
13
c nmr(75mhz,cdcl3)δ160.4,150.9,140.7,137.5,133.5,133.0,131.6,129.0,128.8,128.7,128.6,127.7,126.4,125.4,122.0,117.9,61.5,13.8。
[0064]
hrms(esi
+
)m/z 394.4722。[m+h]
+
。
[0065]
实施例2
[0066]
(e)-3-(4-甲氧基苯基)-1-苯基-5-苯乙烯基-1h-吡唑-4-甲酸乙酯(iii-2),具体结构式如下:
[0067][0068]
合成方法具体如下:
[0069]
以4-甲氧基n-苯基苯并腙酰氯(26mg,0.1mmol)替代n-苯基苯并腙酰氯为原料,其余同实施例1,得黄色油状液体iii-2号27mg,收率65%。
[0070]
对本实施例中的iii-2号物质进行进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0071]1h nmr(400mhz,chloroform-d)δ7.70
–
7.66(m,2h),7.54
–
7.32(m,11h),7.02
–
6.97(m,2h),6.91(d,j=16.6hz,1h),4.27(q,j=7.1hz,2h),3.88(s,3h),1.17(t,j=7.1hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.4,159.8,150.6,140.7,137.5,133.3,131.5,130.2,128.8,128.6,128.3,127.7,126.4,125.4,125.3,121.7,118.1,114.0,61.4,55.3,13.8。
[0072]
hrms(esi+)m/z 424.2525。[m+h]
+
。
[0073]
实施例3
[0074]
(e)-3-(3,4-二甲氧基苯基)-1-苯基-5-苯乙烯基-1h-吡唑-4-甲酸乙酯(iii-3),具体结构式如下:
[0075][0076]
合成方法具体如下:
[0077]
以3,4-二甲氧基n-苯基苯并腙酰氯(29mg,0.1mmol)替代n-苯基苯并腙酰氯为原料,其余同实施例1,得黄色油状液体iii-3号23mg,收率50%。
[0078]
对本实施例中的iii-3号物质进行进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0079]1h nmr(400mhz,chloroform-d)δ7.53
–
7.30(m,12h),7.28
–
7.23(m,1h),6.99
–
6.88(m,2h),4.27(q,j=7.1hz,2h),3.96(s,3h),3.89(s,3h),1.17(t,j=7.1hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.4,150.6,149.2,148.9,140.7,137.4,133.4,131.5,128.8,128.7,128.4,127.8,126.3,125.5,125.4,121.7,121.6,118.0,111.9,111.1,61.5,56.0,55.9,13.9。
[0080]
hrms(esi+)m/z 454.2539。[m+h]
+
。
[0081]
实施例4
[0082]
(e)-1-苯基-5-苯乙烯基-3-(3,4,5-三甲氧基苯基)-1h-吡唑-4-羧酸乙酯(iii-4),具体结构式如下:
[0083][0084]
合成方法具体如下:
[0085]
以3,4,5-三甲氧基n-苯基苯并腙酰氯(32mg,0.1mmol)替代n-苯基苯并腙酰氯为原料,其余同实施例1,得黄色油状液体iii-4号18mg,收率37%。
[0086]
对本实施例中的iii-4号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0087]1h nmr(400mhz,chloroform-d)δ7.55
–
7.31(m,11h),7.29
–
7.26(m,1h),6.99(s,2h),4.27(q,j=7.2hz,2h),3.92(s,3h),3.85(s,6h),1.17(t,j=7.2hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.3,153.3,150.6,140.6,138.2,137.3,133.7,128.8,128.7,128.5,127.8,126.3,125.5,121.8,117.8,106.0,103.8,61.5,61.0,56.2,13.8。
[0088]
hrms(esi+)m/z 484.2523。[m+h]
+
。
[0089]
实施例5
[0090]
(e)-3-(4-氟苯基)-1-苯基-5-苯乙烯基-1h-吡唑-4-甲酸乙酯(iii-5),具体结构式如下:
[0091][0092]
合成方法具体如下:
[0093]
以4-氟n-苯基苯并腙酰氯(24.8mg,0.1mmol)替代n-苯基苯并腙酰氯为原料,其余
同实施例1,得黄色油状液体iii-5号26mg,收率63%。
[0094]
对本实施例中的iii-5号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0095]1h nmr(300mhz,chloroform-d)δ7.73(ddd,j=8.6,5.4,2.6hz,2h),7.52
–
7.32(m,11h),7.20
–
7.10(m,2h),6.84(d,j=16.7hz,1h),4.28(q,j=7.1hz,2h),1.18(t,j=7.1hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.26,153.33,150.56,140.63,138.27,137.31,133.68,131.62,128.79,128.73,128.50,128.35,127.83,126.28,125.45,121.80,117.79,106.10,61.49,60.99,56.18,13.85。
[0096]
hrms(esi+)m/z 412.1608。[m+h]
+
。
[0097]
实施例6
[0098]
(e)-3-(4-氯苯基)-1-苯基-5-苯乙烯基-1h-吡唑-4-甲酸乙酯(iii-6),具体结构式如下:
[0099]
合成方法具体如下:
[0100]
以4-氯n-苯基苯并腙酰氯(26.4mg,0.1mmol)替代n-苯基苯并腙酰氯为原料,其余同实施例1,得黄色油状液体iii-6号26mg,收率60%。
[0101]
对本实施例中的iii-6号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0102]1h nmr(400mhz,chloroform-d)δ7.77
–
7.65(m,2h),7.52
–
7.34(m,13h),6.86(d,j=16.7hz,1h),4.27(q,j=7.1hz,2h),1.17(t,j=7.1hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.2,149.6,140.6,137.2,134.4,133.9,131.4,130.2,128.8,128.8,128.7,128.6,127.9,126.4,125.4,121.9,117.6,61.5,13.8。
[0103]
hrms(esi+)m/z 428.1316。[m+h]
+
。
[0104]
实施例7
[0105]
(e)-3-(4-溴苯基)-1-苯基-5-苯乙烯基-1h-吡唑-4-甲酸乙酯(iii-7),具体结构式如下:
[0106][0107]
合成方法具体如下:
[0108]
以4-氯n-苯基苯并腙酰氯(30mg,0.1mmol)替代n-苯基苯并腙酰氯为原料,其余同实施例1,得黄色油状液体iii-7号32mg,收率69%。
[0109]
对本实施例中的iii-7号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0110]1h nmr(400mhz,chloroform-d)δ7.56
–
7.47(m,5h),7.43
–
7.34(m,8h),7.30(td,j=3.3,1.8hz,1h),7.27
–
7.21(m,1h),6.85(d,j=16.6hz,1h),4.27(q,j=7.1hz,2h),1.17(t,j=7.1hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.1,149.3,137.2,134.7,134.5,134.0,129.8,128.9,128.8,128.7,128.6,128.5,127.9,127.1,126.9,126.4,125.4,122.0,117.5,61.6,13.8。
[0111]
hrms(esi+)m/z 472.0816。[m+h]
+
。
[0112]
实施例8
[0113]
(e)-3-(3-氯-4-氟苯基)-1-苯基-5-苯乙烯基-1h-吡唑-4-甲酸乙酯(iii-8),具体结构式如下:
[0114][0115]
合成方法具体如下:
[0116]
以3-氯-4-氟n-苯基苯并腙酰氯(28mg,0.1mmol)替代n-苯基苯并腙酰氯为原料,其余同实施例1,得黄色油状液体iii-8号26mg,收率58%。
[0117]
对本实施例中的iii-8号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0118]1h nmr(400mhz,chloroform-d)δ7.86(dd,j=7.1,2.1hz,1h),7.63(ddd,j=8.5,4.6,2.2hz,1h),7.53
–
7.46(m,5h),7.44
–
7.33(m,6h),7.22(t,j=8.7hz,1h),6.90
–
6.80(m,1h),4.27(q,j=7.1hz,2h),1.17(t,j=7.1hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.0,156.9,148.5,140.5,137.1,134.1,131.1,130.2,130.2,129.5,128.9,128.8,128.7,128.7,128.0,126.9,126.4,126.2,125.4,121.9,121.4,121.3,117.4,116.8,116.6,61.6,13.8。
[0119]
hrms(esi+)m/z 446.1218。[m+h]
+
。
[0120]
实施例9
[0121]
(e)-5-(4-甲氧基苯乙烯基)-1,3-二苯基-1h-吡唑-4-甲酸乙酯(iii-9),具体结构式如下:
[0122][0123]
合成方法具体如下:
[0124]
以5-乙酰氧基-5-(4-甲氧基苯基)五-2,3-二烯酸乙酯(29mg,0.1mmol)替代5-乙酰氧基-5-苯基五-2,3-二烯酸乙酯为原料,其余同实施例1,得黄色油状液体iii-9号28mg,收率66%。
[0125]
对本实施例中的iii-9号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱
数据。并对其进行质谱分析,获得质谱数据。
[0126]1h nmr(400mhz,chloroform-d)δ7.79
–
7.71(m,2h),7.54
–
7.39(m,8h),7.36
–
7.30(m,2h),7.25(d,j=16.6hz,1h),6.94
–
6.79(m,3h),4.27(q,j=7.2hz,2h),3.84(s,3h),1.17(t,j=7.1hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.4,159.4,150.7,140.7,133.1,133.1,131.2,130.3,129.3,129.0,128.7,128.5,128.3,128.3,127.6,125.4,122.3,115.7,114.1,61.4,55.3,13.8。
[0127]
hrms(esi+)m/z 424.1816。[m+h]
+
。
[0128]
实施例10
[0129]
(e)-5-(4-氯苯乙烯基)-1,3-二苯基-1h-吡唑-4-甲酸乙酯(iii-10),具体结构式如下:
[0130][0131]
合成方法具体如下:
[0132]
以5-乙酰氧基-5-(4-氯苯基)五-2,3-二烯酸乙酯(29mg,0.1mmol)替代5-乙酰氧基-5-苯基五-2,3-二烯酸乙酯为原料,其余同实施例1,得黄色油状液体iii-9号32mg,收率75%。
[0133]
对本实施例中的iii-10号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0134]1h nmr(400mhz,chloroform-d)1h nmr(400mhz,chloroform-d)δ7.77
–
7.67(m,2h),7.57
–
7.40(m,9h),7.33(d,j=15.9hz,4h),6.82(d,j=16.7hz,1h),4.27(q,j=7.1hz,2h),1.15(t,j=7.2hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.3,150.9,140.7,135.9,133.3,132.9,132.1,131.6,129.4,129.2,129.0,128.8,128.8,128.6,128.5,128.0,127.8,127.5,126.2,125.4,121.7,118.5,61.5,13.8。
[0135]
hrms(esi+)m/z 428.1314。[m+h]
+
。
[0136]
实施例11
[0137]
(e)-5-(4-氟苯乙烯基)-1,3-二苯基-1h-吡唑-4-甲酸乙酯(iii-11),具体结构式如下:
[0138][0139]
合成方法具体如下:
[0140]
以5-乙酰氧基-5-(4-氟苯基)五-2,3-二烯酸乙酯(28mg,0.1mmol)替代5-乙酰氧基-5-苯基五-2,3-二烯酸乙酯为原料,其余同实施例1,得黄色油状液体iii-10号27mg,收率65%。
[0141]
对本实施例中的iii-11号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱
数据。并对其进行质谱分析,获得质谱数据。
[0142]1h nmr(400mhz,chloroform-d)δ7.79
–
7.66(m,2h),7.57
–
7.27(m,11h),7.03(t,j=8.7hz,2h),6.83(d,j=16.6hz,1h),4.27(q,j=7.1hz,2h),1.16(t,j=7.1hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.3,140.7,133.6,132.9,132.3,131.5,129.0,128.8,128.6,128.4,128.4,127.9,127.8,125.4,121.8,117.6,115.7,115.5,61.5,13.8。
[0143]
hrms(esi+)m/z 412.1634。[m+h]
+
。
[0144]
实施例12
[0145]
(e)-5-(4-甲基苯乙烯基)-1,3-二苯基-1h-吡唑-4-甲酸乙酯(iii-12),具体结构式如下:
[0146][0147]
合成方法具体如下:
[0148]
以5-乙酰氧基-5-(4-甲基苯基)五-2,3-二烯酸乙酯(27mg,0.1mmol)替代5-乙酰氧基-5-苯基五-2,3-二烯酸乙酯为原料,其余同实施例1,得黄色油状液体iii-12号16mg,收率59%。
[0149]
对本实施例中的iii-12号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0150]1h nmr(400mhz,chloroform-d)δ7.79
–
7.72(m,2h),7.53
–
7.41(m,8h),7.30(dd,j=10.8,2.7hz,3h),7.16(d,j=7.9hz,2h),6.84(d,j=16.7hz,1h),4.27(q,j=7.1hz,2h),2.37(s,3h),1.18(t,j=7.1hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.4,150.8,140.7,137.7,134.7,133.5,133.0,131.4,129.3,129.0,128.8,128.5,128.4,128.3,126.3,125.4,122.1,116.8,61.4,21.3,13.8。
[0151]
hrms(esi+)m/z 408.1871。[m+h]
+
。
[0152]
实施例13
[0153]
(e)-5-(4-异丙基苯乙烯基)-1,3-二苯基-1h-吡唑-4-甲酸乙酯(iii-13),具体结构式如下:
[0154][0155]
合成方法具体如下:
[0156]
以5-乙酰氧基-5-(4-异丙基苯基)五-2,3-二烯酸乙酯(30mg,0.1mmol)替代5-乙酰氧基-5-苯基五-2,3-二烯酸乙酯为原料,其余同实施例1,得黄色油状液体iii-13号27mg,收率62%。
[0157]
对本实施例中的iii-13号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0158]1h nmr(400mhz,chloroform-d)δ7.79
–
7.69(m,2h),7.54
–
7.42(m,8h),7.38
–
7.30(m,3h),7.21(d,j=8.3hz,2h),6.85(d,j=16.7hz,1h),4.28(qd,j=7.2,2.5hz,2h),2.93(p,j=6.9hz,1h),1.29(s,3h),1.27(s,3h),1.19(t,j=7.1hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.4,150.8,148.7,140.7,135.1,133.5,133.0,131.4,129.0,128.8,128.5,128.4,128.3,126.7,126.4,125.4,122.1,117.0,61.4,33.9,23.9,13.9。
[0159]
hrms(esi+)m/z 436.2387。[m+h]
+
。
[0160]
实施例14
[0161]
(e)-5-(2-氯苯乙烯基)-1,3-二苯基-1h-吡唑-4-甲酸乙酯(iii-14),具体结构式如下:
[0162][0163]
合成方法具体如下:
[0164]
以5-乙酰氧基-5-(2-氯苯基)五-2,3-二烯酸乙酯(30mg,0.1mmol)替代5-乙酰氧基-5-苯基五-2,3-二烯酸乙酯为原料,其余同实施例1,得黄色油状液体iii-14号27mg,收率63%。
[0165]
对本实施例中的iii-14号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0166]1h nmr(400mhz,chloroform-d)δ7.75
–
7.67(m,2h),7.58
–
7.35(m,10h),7.30
–
7.26(m,2h),7.24
–
7.20(m,1h),6.80(d,j=16.7hz,1h),4.27(q,j=7.1hz,2h),1.16(t,j=7.1hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.2,139.3,135.5,134.6,132.8,131.9,129.8,129.5,129.2,129.0,128.8,128.6,128.5,127.8,127.6,126.2,126.2,125.4,124.5,119.4,61.5,13.8。
[0167]
hrms(esi+)m/z 428.1314。[m+h]
+
。
[0168]
实施例15
[0169]
(e)-5-(4-溴苯乙烯基)-1,3-二苯基-1h-吡唑-4-甲酸乙酯(iii-15),具体结构式如下:
[0170][0171]
合成方法具体如下:
[0172]
以5-乙酰氧基-5-(4-溴苯基)五-2,3-二烯酸乙酯(34mg,0.1mmol)替代5-乙酰氧基-5-苯基五-2,3-二烯酸乙酯为原料,其余同实施例1,得黄色油状液体iii-15号28mg,收率60%。
[0173]
对本实施例中的iii-15号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0174]1h nmr(400mhz,chloroform-d)δ7.76
–
7.67(m,2h),7.54
–
7.35(m,11h),7.27
–
7.19(m,2h),6.80(d,j=16.7hz,1h),4.26(q,j=7.1hz,2h),1.15(t,j=7.1hz,3h)。
13
c nmr(101mhz,cdcl3)δ162.3,152.5,142.5,140.4,134.7,134.1,133.8,132.0,129.7,129.6,128.8,128.3,127.6,122.4,121.7,116.1,105.1,60.7,13.8。
[0175]
hrms(esi+)m/z 472.1813。[m+h]
+
。
[0176]
实施例16
[0177]
(e)-1,3-二苯基-5-(2-(噻吩-2-基)乙烯基)-1h-吡唑-4-甲酸乙酯(iii-16),具体结构式如下:
[0178][0179]
合成方法具体如下:
[0180]
以5-乙酰氧基-5-(噻吩-2-基)五-2,3-二烯酸乙酯(27mg,0.1mmol)替代5-乙酰氧基-5-苯基五-2,3-二烯酸乙酯为原料,其余同实施例1,得黄色油状液体iii-16号15mg,收率38%。
[0181]
对本实施例中的iii-16号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0182]1h nmr(400mhz,chloroform-d)1h nmr(400mhz,chloroform-d)1h nmr(400mhz,chloroform-d)1h nmr(400mhz,chloroform-d)δ7.79
–
7.71(m,2h),7.53
–
7.41(m,8h),7.29
–
7.19(m,2h),7.03
–
6.89(m,3h),4.29(q,j=7.1hz,2h),1.22(t,j=7.2hz,3h)。
13
c nmr(101mhz,cdcl3)δ160.2,150.7,143.0,140.6,132.9,131.3,129.0,128.8,128.6,128.4,127.6,126.5,126.2,125.4,124.5,121.5,117.5,61.5,13.8。
[0183]
hrms(esi+)m/z 400.1276。[m+h]
+
。
[0184]
实施例17
[0185]
(e)-n-异丙基-1,3-二苯基-5-苯乙烯基-1h-吡唑-4-甲酰胺(
ⅳ‑
1),具体结构式如下:
[0186][0187]
合成方法具体如下:
[0188]
采用实施例1所制备的(e)-1,3-二苯基-5-苯乙烯基-1h-吡唑-4-羧酸乙酯(iii-1)进行制备,具体步骤为:
[0189]
在150ml三颈瓶中加入(e)-1,3-二苯基-5-苯乙烯基-1h-吡唑-4-羧酸乙酯和氢氧化锂,将其溶解于四氢呋喃水溶液中,得到衍生物混合液,该四氢呋喃水溶液采用体积比为
1:1的四氢呋喃和去离子水配制。将衍生物混合液油浴锅中110℃加热回流5h,进行水解反应,用薄层色谱法监测反应,直至(e)-1,3-二苯基-5-苯乙烯基-1h-吡唑-4-羧酸乙酯全部反应;
[0190]
反应完成后,将四氢呋喃真空旋干,获得第二旋干物,在第二旋干物中加入稀盐酸调节ph至2,析出黄色固体,然后通过真空过滤收集产物并干燥以获得水解产物a;
[0191]
将水解产物a(36.6mg,0.1mmol)和亚硫酰氯(47.2mg,0.4mmol)溶解在chcl3(10ml)中。然后在80℃回流,当水解产物a被消耗完毕时(通过tlc监测),将混合物浓缩,得相应的酰氯化合物b。将酰氯化合物b(38.4mg,0.1mmol)溶解在二氯甲烷(2ml)中,并向其中滴加三乙胺(0.05ml)和2-丙胺(6mg,0.1mmol)的混合溶液。保持搅拌,将上述体系置于氩气保护下,并于0℃下进行胺化反应。反应结束后,在反应混合物中加入200-300目硅胶,旋干溶剂制沙,获得第三旋干物,随后通过pe:ea=5:1-1:1的柱色谱法对第三旋干物进行纯化,分离得到相应的白色固体
ⅳ‑
1号22.4mg,收率55%。
[0192]
对本实施例中的
ⅳ‑
1号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0193]1h nmr(300mhz,chloroform-d)δ7.76
–
7.70(m,2h),7.68
–
7.62(m,2h),7.53
–
7.29(m,11h),7.26(d,j=7.1hz,1h),7.11(d,j=16.6hz,1h),6.94(d,j=16.6hz,1h),4.24(dp,j=8.1,6.5hz,1h),1.12(d,j=6.6hz,6h)。
13
c nmr(101mhz,cdcl3)δ160.4,151.1,139.5,137.3,135.6,132.7,132.4,129.2,128.8,128.7,128.6,128.4,128.2,127.7,126.3,124.0,118.2,117.4,42.2,22.3。
[0194]
hrms(esi+)m/z 407.2025。[m+h]
+
。
[0195]
实施例18
[0196]
(e)-n-(2-羟乙基)-1,3-二苯基-5-苯乙烯基-1h-吡唑-4-甲酰胺(
ⅳ‑
2),具体结构式如下:
[0197][0198]
合成方法具体如下:
[0199]
以乙醇胺(6mg,0.1mmol)替代2-丙胺为原料,其余同实施例9,得黄色固体
ⅳ‑
2号27mg,收率67%。
[0200]
对本实施例中的
ⅳ‑
2号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0201]1h nmr(300mhz,chloroform-d)δ7.77
–
7.70(m,2h),7.68
–
7.61(m,2h),7.55
–
7.29(m,11h),7.27(d,j=7.7hz,1h),7.12(d,j=16.5hz,1h),6.94(d,j=16.6hz,1h),3.66(dd,j=5.6,4.3hz,2h),3.52(td,j=5.7,4.2hz,2h)。
13
c nmr(101mhz,cdcl3)δ162.0,139.6,137.2,135.2,132.6,129.2,128.7,128.7,128.6,128.4,128.3,127.8,126.3,
124.1,118.6,117.4,61.4,53.5。
[0202]
hrms(esi+)m/z 409.1825。[m+h]
+
。
[0203]
实施例19
[0204]
(e)-(1,3-二苯基-5-苯乙烯基-1h-吡唑-4-羰基)甘氨酸乙酯(
ⅳ‑
3),具体结构式如下:
[0205][0206]
合成方法具体如下:
[0207]
以甘氨酸乙酯盐酸盐(10.3mg,0.1mmol)替代2-丙胺为原料,其余同实施例9,得黄色固体
ⅳ‑
3号22mg,收率49%。
[0208]
对本实施例中的
ⅳ‑
3号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0209]1h nmr(300mhz,chloroform-d)δ7.78
–
7.70(m,2h),7.67
–
7.59(m,2h),7.53
–
7.29(m,12h),7.17(d,j=16.6hz,1h),6.92(d,j=16.6hz,1h),4.23
–
4.17(m,2h),4.16
–
4.12(m,2h),1.27(d,j=7.2hz,3h)。
13
c nmr(101mhz,cdcl3)δ169.0,161.1,151.0,139.6,137.2,134.4,133.3,132.7,129.2,128.8,128.7,128.4,128.3,127.8,126.5,124.3,119.0,117.3,61.8,41.7,14.1。
[0210]
hrms(esi+)m/z 451.1945。[m+h]
+
。
[0211]
实施例20
[0212]
(e)-(1,3-二苯基-5-苯乙烯基-1h-吡唑-4-羰基)苯丙氨酸乙酯(
ⅳ‑
4),具体结构式如下:
[0213][0214]
合成方法具体如下:
[0215]
以苯丙氨酸乙酯盐酸盐(19.3mg,0.1mmol)替代2-丙胺为原料,其余同实施例9,得黄色固体
ⅳ‑
4号20mg,收率37%。
[0216]
对本实施例中的
ⅳ‑
4号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0217]1h nmr(300mhz,chloroform-d)δ7.75
–
7.69(m,2h),7.62
–
7.56(m,2h),7.53
–
7.30
(m,13h),7.27
–
7.22(m,1h),7.18(dd,j=5.0,1.9hz,3h),6.93
–
6.89(m,2h),5.03(dt,j=8.0,5.8hz,1h),4.13(qt,j=7.1,3.7hz,2h),3.22
–
3.02(m,2h),1.19(t,j=7.1hz,3h)。
13
c nmr(101mhz,cdcl3)δ170.7,160.7,151.0,139.6,137.2,135.3,134.7,133.4,132.7,129.2,128.8,128.7,128.6,128.3,127.8,127.3,126.5,124.4,119.0,117.2,61.8,53.5,37.7,14.1。
[0218]
hrms(esi+)m/z 541.2443。[m+h]
+
。
[0219]
实施例21
[0220]
(e)-n,1,3-三苯基-5-苯乙烯基-1h-吡唑-4-甲酰胺(
ⅳ‑
5),具体结构式如下:
[0221][0222]
合成方法具体如下:
[0223]
以苯胺(9mg,0.1mmol)替代2-丙胺为原料,其余同实施例9,得黄色固体
ⅳ‑
5号21mg,收率48%。
[0224]
对本实施例中的
ⅳ‑
5号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0225]1h nmr(300mhz,chloroform-d)δ7.79
–
7.73(m,2h),7.71
–
7.64(m,2h),7.60(s,1h),7.53
–
7.29(m,15h),7.27
–
7.15(m,2h),6.98(d,j=16.6hz,1h)。
13
c nmr(101mhz,cdcl3)δ158.9,151.2,139.6,137.0,135.0,133.5,132.6,129.3,129.2,128.8,128.7,128.7,128.5,128.4,128.0,126.5,125.3,124.3,120.2,119.0,117.3。
[0226]
hrms(esi+)m/z 441.1823。[m+h]
+
。
[0227]
实施例22
[0228]
(e)-1,3-二苯基-5-苯乙烯基-n-(对甲苯基)-1h-吡唑-4-甲酰胺(
ⅳ‑
6),具体结构式如下:
[0229][0230]
合成方法具体如下:
[0231]
以4-甲基苯胺(10.8mg,0.1mmol)替代2-丙胺为原料,其余同实施例9,得黄色固体
ⅳ‑
5号29mg,收率64%。
[0232]
对本实施例中的
ⅳ‑
6号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0233]1h nmr(300mhz,chloroform-d)δ7.76(dd,j=7.9,1.7hz,2h),7.71
–
7.65(m,2h),7.55
–
7.35(m,10h),7.35
–
7.29(m,4h),7.25(d,j=15.2hz,1h),7.17
–
7.11(m,2h),6.97(d,j=16.6hz,1h),2.34(s,3h)。
13
c nmr(101mhz,cdcl3)δ158.7,151.2,139.6,137.0,135.1,134.4,133.4,132.6,129.7,129.3,128.8,128.7,128.7,128.5,128.4,127.9,126.4,124.2,120.3,118.8,117.3,20.9。
[0234]
hrms(esi+)m/z 455.2055。[m+h]
+
。
[0235]
实施例23
[0236]
(e)-n-(4-氯-3-氟)-1,3-二苯基-5-苯乙烯基-1h-吡唑-4-甲酰胺(
ⅳ‑
7),具体结构式如下:
[0237]
合成方法具体如下:
[0238][0239]
合成方法具体如下:
[0240]
以4-氯-3-氟苯胺(14.5mg,0.1mmol)替代2-丙胺为原料,其余同实施例9,得黄色固体
ⅳ‑
7号20mg,收率40%。
[0241]
对本实施例中的
ⅳ‑
7号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0242]1h nmr(300mhz,chloroform-d)δ7.79
–
7.72(m,2h),7.70
–
7.61(m,3h),7.56
–
7.30(m,13h),7.17
–
7.12(m,1h),7.08(d,j=8.5hz,1h),6.95(d,j=16.6hz,1h)。
13
c nmr(101mhz,cdcl3)δ158.8,154.1,151.2,139.5,136.8,134.4,134.0,133.6,132.3,129.5,129.3,128.8,128.7,128.6,128.2,127.5,126.4,124.3,122.5,121.5,121.3,119.9,119.8,119.3,117.2,116.9,116.7。
[0243]
hrms(esi+)m/z 493.1401。[m+h]
+
。
[0244]
实施例24
[0245]
(e)-n-(3,5-二甲氧基苯基)-1,3-二苯基-5-苯乙烯基-1h-吡唑-4-甲酰胺(
ⅳ‑
8),具体结构式如下:
[0246][0247]
合成方法具体如下:
[0248]
以3,5-二甲氧基苯胺(15.3mg,0.1mmol)替代2-丙胺为原料,其余同实施例9,得黄色固体
ⅳ‑
8号27mg,收率54%。
[0249]
对本实施例中的
ⅳ‑
8号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0250]1h nmr(300mhz,chloroform-d)δ7.76(dd,j=7.9,1.7hz,2h),7.70
–
7.62(m,2h),7.59(s,1h),7.55
–
7.29(m,12h),7.19(d,j=16.6hz,1h),6.96(d,j=16.6hz,1h),6.63(d,j=2.2hz,2h),3.72(s,6h)。
13
c nmr(101mhz,cdcl3)δ161.1,158.7,151.1,139.5,138.7,137.0,134.9,133.8,132.5,129.3,128.7,128.7,128.5,128.4,128.0,126.5,124.3,119.0,117.4,98.4,97.7,55.4。
[0251]
hrms(esi+)m/z 501.2132。[m+h]
+
。
[0252]
实施例25
[0253]
(e)-(1,3-二苯基-5-苯乙烯基-1h-吡唑-4-基)(吗啉)甲酮(
ⅳ‑
9),具体结构式如下:
[0254][0255]
合成方法具体如下:
[0256]
以吗啉(9mg,0.1mmol)替代2-丙胺为原料,其余同实施例9,得黄色固体
ⅳ‑
9号5mg,收率11%。
[0257]
对本实施例中的
ⅳ‑
9号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0258]1h nmr(300mhz,chloroform-d)δ7.79
–
7.73(m,2h),7.72
–
7.65(m,2h),7.56
–
7.30(m,12h),6.98(q,j=16.5hz,2h),3.83
–
3.63(m,3h),3.57
–
3.39(m,2h),3.27(ddd,j=13.0,6.9,3.1hz,1h),3.13
–
2.96(m,2h)。
13
c nmr(101mhz,cdcl3)δ161.9,151.5,139.4,137.3,133.6,132.5,131.3,129.5,128.7,128.7,128.5,128.2,127.8,126.4,123.1,
117.9,117.4,66.3,46.8。
[0259]
hrms(esi+)m/z 435.1915。[m+h]
+
。
[0260]
实施例26
[0261]
(e)-(1,3-二苯基-5-苯乙烯基-1h-吡唑-4-基)(4-甲基吡嗪-1-基)甲酮(
ⅳ‑
10),具体结构式如下:
[0262][0263]
合成方法具体如下:
[0264]
以1-甲基哌嗪(10mg,0.1mmol)替代2-丙胺为原料,其余同实施例9,得黄色固体
ⅳ‑
10号12mg,收率28%。
[0265]
对本实施例中的
ⅳ‑
10号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0266]1h nmr(300mhz,chloroform-d)δ7.80
–
7.73(m,2h),7.71
–
7.65(m,2h),7.55
–
7.29(m,11h),7.08
–
6.85(m,2h),3.79(d,j=5.5hz,2h),3.20(dddd,j=51.8,13.1,6.9,3.2hz,2h),2.41(dq,j=12.3,7.2,6.1hz,1h),2.33
–
2.18(m,2h),2.16(s,3h),1.78(d,j=7.7hz,1h)。
13
c nmr(101mhz,cdcl3)δ161.7,151.5,139.4,137.4,134.0,132.6,131.2,129.4,128.7,128.7,128.4,128.0,127.7,126.4,123.1,117.7,117.5,54.5,45.8,41.8。
[0267]
hrms(esi+)m/z 448.2312。[m+h]
+
。
[0268]
实施例27
[0269]
(e)-(1,3-二苯基-5-苯乙烯基-1h-吡唑-4-基)(4-乙基吡嗪-1-基)甲酮(
ⅳ‑
11),具体结构式如下:
[0270][0271]
合成方法具体如下:
[0272]
以1-乙基哌嗪(11.4mg,0.1mmol)替代2-丙胺为原料,其余同实施例9,得黄色固体
ⅳ‑
11号5mg,收率10%。
[0273]
对本实施例中的
ⅳ‑
11号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0274]1h nmr(300mhz,chloroform-d)δ7.80
–
7.73(m,2h),7.70
–
7.64(m,2h),7.53
–
7.31
(m,11h),7.03(d,j=16.5hz,1h),6.90(d,j=16.6hz,1h),3.80(s,2h),3.38
–
3.08(m,2h),2.46(q,j=5.5,5.0hz,1h),2.30(q,j=7.2hz,4h),1.29
–
1.26(m,1h),1.00(t,j=7.2hz,3h)。
13
c nmr(101mhz,cdcl3)δ161.7,151.5,139.4,137.4,134.0,132.6,131.1,129.4,128.7,128.7,128.7,128.4,128.0,127.7,126.4,123.1,117.5,52.0,51.8,46.3,11.7。
[0275]
hrms(esi+)m/z 462.2450。[m+h]
+
。
[0276]
实施例28
[0277]
(e)-(3-(3,4-二甲氧基苯基)-1-苯基-5-苯乙烯基-1h-吡唑-4-基)(4甲基哌嗪-1-基)甲酮(
ⅳ‑
12),具体结构式如下:
[0278][0279]
合成方法具体如下:
[0280]
以iii-3(45.4mg,0.1mmol)替代iii-1,并以1-乙基哌嗪(10mg,0.1mmol)替代2-丙胺为原料,其余同实施例9,得黄色固体
ⅳ‑
12号13mg,收率25%。
[0281]
对本实施例中的
ⅳ‑
12号物质进行核磁共振波谱检测,获得核磁氢谱和核磁碳谱数据。并对其进行质谱分析,获得质谱数据。
[0282]1h nmr(400mhz,chloroform-d)δ7.70
–
7.64(m,2h),7.55
–
7.29(m,9h),7.28
–
7.24(m,1h),7.07
–
6.87(m,3h),3.96(s,3h),3.94(s,3h),3.80(d,j=32.9hz,2h),3.26(d,j=61.4hz,2h),2.38(d,j=66.9hz,3h),2.18(s,3h),2.07(s,1h)。
13
c nmr(101mhz,cdcl3)δ161.7,151.3,149.4,149.1,139.4,137.3,131.2,129.5,128.8,128.0,127.8,126.3,125.2,123.1,121.4,117.6,111.6,111.2,56.0,53.9,14.2。
[0283]
hrms(esi+)m/z 508.2520。[m+h]
+
。
[0284]
实施例29
[0285]
1,3,5-三取代-吡唑-4-羧酸衍生物在细胞活性分析中的应用。该应用具体为(以iii-1为例):
[0286]
(1)接种细胞:用含10%胎牛血清的培养基制备单细胞悬液,每孔接种1000个du145细胞至96孔板中,每孔体积100ul;
[0287]
(2)配药:将iii-1号(1mg,2.5μmol)置于1mldmso中溶解,配制成2500μm的第一羧酸衍生物溶液。随后在第一羧酸衍生物溶液中加入9ml ldmso将其稀释10倍,配置成250μm的第二羧酸衍生物溶液。接着再取1ml稀释后的250μm的第二羧酸衍生物溶液,加入4mldmso将其稀释5倍,配制成50μm的第三羧酸衍生物溶液,留存待用。取2ml 50μm的第三羧酸衍生物溶液,加入3ml dms0将其稀释2.5倍,配制成20μm的第四羧酸衍生物溶液,留存待用。取1m 20μm的第四羧酸衍生物溶液,加入1ml dms0将其稀释2倍,配制成10μm的第五羧酸衍生物溶液,留存待用。
[0288]
(3)给药:细胞接种后24小时,每孔分别加入100ul第五羧酸衍生物溶液、第四羧酸
衍生物溶液和第三羧酸衍生物溶液,孵育4天;
[0289]
(4)着色:培养4天后,每孔加入10ul mtt溶液(5mg/ml in pbs,ph=7.4);继续孵育4h终止培养,吸弃孔内培养上清,将悬浮细胞离心后吸弃孔内培养上清;每孔加入100ul dmso,摇晃10分钟,使晶体完全溶解;
[0290]
(5)比色法:选择570nm波长,在酶联免疫吸附仪上测量各孔的吸光值,记录结果,用graphpad软件处理数据,列入下表1,得到ic
50
。
[0291]
表1
[0292]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1