多能干细胞制造系统和生产诱导多能干细胞的方法与流程
多能干细胞制造系统和生产诱导多能干细胞的方法
1.本技术是向中国国家知识产权局提交的申请日为2016年8月30日的发明名称为“多能干细胞制造系统和生产诱导多能干细胞的方法”的第201680050145.x号申请的分案申请。
技术领域
2.本发明涉及一种细胞技术,涉及一种多能干细胞制造系统、一种诱导干细胞的方法、一种用于干细胞的漂浮培养方法、一种用于干细胞的漂浮培养容器、一种生产诱导多能干细胞的方法,以及一种从动物细胞生产特定体细胞的方法。
背景技术:
3.胚胎干细胞(es细胞)是从人或小鼠早期胚胎建立的干细胞。es细胞表现出多能性,这种多能性允许它们分化成它们源自的生物体中的每一种细胞。目前,在细胞移植治疗中利用人es细胞来治疗许多疾病,包括帕金森病、幼年型糖尿病和白血病。然而,es细胞的移植存在缺点。值得注意的是,es细胞的移植可能以类似于不成功的器官移植之后发生的排斥的方式触发免疫排斥。此外,使用通过破坏人胚胎所建立的es细胞已引起大量基于伦理的批评和强烈的反对。
4.在这些情况的背景下,京都大学教授shinya yamanaka通过将oct3/4、klf4、c-myc和sox2四种基因转移到体细胞中,成功建立了诱导多能干细胞(ips细胞)。为此,他被授予2012年诺贝尔生理学或医学奖(参见例如专利文献1)。ips细胞是理想的多能细胞类型,因为它们避免了免疫排斥和伦理问题两者。因此,预期ips细胞将用于细胞移植治疗。
5.(诱导干细胞的方法、用于干细胞的漂浮培养方法和用于干细胞的漂浮培养容器的背景技术)
6.诱导多能干(ips)细胞具有两种特征性的潜能。首先是生成体内所有体细胞的潜能。其次是半永久增殖的能力。因为ips细胞表现出这两种潜能,所以通过从个体自身体细胞生产ips细胞并将这些细胞转化为感兴趣的体细胞,它们将用于移植治疗而无排斥。因此,ips细胞在再生医学领域具有巨大的前景。
7.(生产诱导多能干细胞的方法的背景技术)
8.诱导多能干(ips)细胞具有两种特征性的潜能。首先是生成体内所有体细胞的潜能。其次是半永久增殖的能力。因为ips细胞表现出这两种潜能,所以它们可以用于移植治疗而无排斥。这可以通过从个体自身体细胞生产ips细胞并将这些细胞转化为感兴趣的体细胞来实现。因此,ips细胞在再生医学领域具有巨大的前景。
9.目前已建立了若干种生产ips细胞的方法。生产ips细胞的方法的典型示例包括使用逆转录病毒或慢病毒的方法,以及使用游离型载体的方法。
10.将描述使用逆转录病毒或慢病毒的方法。逆转录病毒或慢病毒可感染体细胞,使得编码重编程因子的基因转移到细胞中。另外,逆转录病毒或慢病毒可将重编程因子插入体细胞的基因组中以诱导重编程因子在细胞中的稳定表达。
11.然而,依赖于使用逆转录病毒或慢病毒的方法是有问题的。首先,将重编程因子插入到体细胞的基因组损伤了现有基因或启动子,并且因此可触发细胞的肿瘤发生。其次,插入到基因组中的重编程因子可在ips细胞转化为体细胞之后再活化。因此,用于移植的源自ips细胞的细胞具有肿瘤发生的风险。事实上,已确认,转移的重编程因子在小鼠模型的体细胞中再活化,并且细胞变成癌性的(参见例如非专利文献1)。
12.此外,使用逆转录病毒或慢病毒生产的ips细胞可能保留残余病毒。当将此类ips细胞移植给患者时,残余病毒可感染患者。因此,这些ips细胞不可用于移植。作为参考,由于对x连锁联合免疫缺陷病(x-scid)进行基因治疗,其中通过逆转录病毒载体将γc基因转移到造血干细胞中,患者已被告知由于载体的插入导致的lm02基因的激活而罹患白血病(参见例如非专利文献2和3)。
13.因此,使用逆转录病毒或慢病毒生产的ips细胞在临床治疗中的使用是有问题的。
14.接下来,将描述使用游离型载体的方法。为了克服使用逆转录病毒或慢病毒的基因转移方法的问题,已经开发了使用游离型载体来生产ips细胞的方法(参见例如非专利文献4)。游离型载体是质粒。游离型载体与细胞分裂同时复制。与逆转录病毒和慢病毒不同,重编程因子未插入到体细胞的基因中。由于这种特征,游离型载体可实现重编程因子的长期细胞内表达以生成ips细胞而无需将基因插入到靶向体细胞的脱氧核糖核酸(dna)中。
15.然而,使用游离型载体的方法也是有问题的。首先,将基因转移到细胞中需要电穿孔,这极大地损伤了细胞;甚至在单个电穿孔事件期间,高百分比的细胞受损。其次,电穿孔不可重复进行。另外,决定使用游离型载体的方法的基因转移效率低于基于逆转录病毒/慢病毒的方法的基因转移效率。
16.最近的研究已揭示,游离型载体的转移可导致载体dna的片段插入到目标ips细胞的基因中。因此,即使当使用游离型载体时,所得ips细胞很有可能将含有已插入到其基因组中的载体片段。因此,此类ips细胞的临床应用仍存在争议。
17.由于这些原因,使用游离型载体生产的ips细胞同样难以在临床上使用。
18.因为如上所述的使用逆转录病毒或慢病毒的方法和使用游离型载体的方法都是有问题的,所以已提出了使用rna来生产ips细胞的方法(参见例如非专利文献6)。然而,尽管已经使用胎儿或新生的成纤维细胞得到成功ips细胞诱导,但没有关于ips细胞使用rna从源自成年人的体细胞的成功诱导的报道。因此,除非可从源自成年人的体细胞生产ips细胞,否则其临床应用是困难的。
19.另外,为了采集生产ips细胞所必需的成纤维细胞,需要收获1平方厘米的皮肤片。这给皮肤供体带来了很大的负担。切除之后,必须通过扩增培养来建立成纤维细胞培养系。随着这些成纤维细胞在扩增过程中增殖,它们很可能将招致基因组损伤和/或染色体畸变。
20.(从动物细胞生产特定体细胞的方法的背景技术)
21.诱导多能干细胞(ips细胞)可生成体内的每一种体细胞。因此,可转化为各种类型的体细胞或组织的ips细胞预期用于细胞移植治疗和药物开发研究。例如,在2014年,从ips细胞生产的视网膜细胞用于移植治疗。世界各地正在进行许多项目来从ips细胞生成脑细胞(和各种其他器官的细胞),以随后用于移植治疗。
22.迄今为止,已经开发出将ips细胞转化为体细胞的各种方法。然而,为了将ips细胞用于移植治疗,诱导ips细胞分化的高效方法是非常重要的。具体而言,需要开发诱导ips细
胞分化成体细胞的仪器,以提高诱导分化的效率和准确性。这种仪器应生产适合移植治疗的功能性体细胞。
23.诱导ips和es细胞分化成体细胞的常规方法依赖于生长因子、激素和/或小分子的各种组合和浓度来操纵细胞的命运,以试图重述自然发育的过程。然而,体内发生的自然发育难以在体外复制,并且效率较低。另外,在人体内,ips细胞至人体细胞的诱导分化相比于在小鼠体内需要更长的时间。例如,生产人成熟神经元细胞需要至少三个月。此外,诱导分化的效率在es/ips细胞系之间大不相同,从而导致诸如诱导体细胞的不均匀性质等问题。当来自同一来源的多个es集落用相同的化学品处理产生了不同的表型时,这证实了这种现象。这些集落中的一些集落分化为脾脏细胞,而其他则成为心脏细胞,从而表明集落之间的分化潜能不同(参见例如非专利文献6)。另外,当尝试使用称为快速再聚集的类胚体聚集体的无血清漂浮培养(sfebq)的方法使大量ips和es细胞类型分化成神经元细胞时,发现尽管ips细胞和es细胞在无神经分化物质的无血清培养基中培养,但一些ips和es集落难以成功转化为神经元细胞(参见例如非专利文献7)。
24.具体而言,已证实,通过使用激素或化学物质的方法从人es/ips细胞诱导分化出的细胞与初始阶段的胎儿体细胞类似。另外,诱导es/ips细胞分化成人成熟体细胞是非常困难的,并且需要数月的长期培养。然而,对于已完成发育的个体的药物发现或医学移植,生产与这些个体的年龄相当的体细胞是至关重要的。
25.神经元细胞包括各种细胞亚型。使用激素或化学物质来诱导es/ips细胞分化成特定神经元亚型的方法未生产同质细胞群。因此,不可实现特定神经元细胞亚型特异性的药物发现筛选。因此,药物发现筛选的有效性低。另外,就医学移植而言,疾病治疗所需的不同的神经元细胞亚型不可富集以用于移植。
26.相比之下,已经提出了一种使用病毒直接将包含用于生成特定体细胞的性质的信息的基因转移到es/ips细胞中来生产感兴趣的体细胞的方法。这种方法使得有可能在比前述依赖于使用激素或化学物质的方法远远更短的时间(两周)内特异性地生产成熟的神经元细胞。例如,通过将特定基因转染到es/ips细胞中可获得同质的兴奋性神经元群体。因此,据认为,可实现特定神经元细胞亚型特异性的药物发现筛选。同样,对于医学移植,可富集和移植特定的神经元细胞亚型以治疗疾病。
27.然而,使用病毒表达特定基因来诱导干细胞分化为体细胞的方法将该基因插入到es/ips细胞的基因组中并损伤了内源基因。因此,不利的是,药物发现筛选不一定准确,并且移植导致肿瘤发生的风险(参见例如非专利文献8和9)。
28.[引文列表]
[0029]
[专利文献]
[0030]
[专利文献1]日本专利no.4183742
[0031]
[非专利文献]
[0032]
[非专利文献1]《自然》,第448卷,313-317页
[0033]
[非专利文献2]《新英格兰医学杂志》,第346卷:1185-1193页,2002年
[0034]
[非专利文献3]《自然》,第302卷:415-419页,2003年
[0035]
[非专利文献4]《自然》,第324卷:797-801页,2009年
[0036]
[非专利文献5]《日本学士院院报》,b辑,《物理和生物科学》,2009年;第85卷(第8
期):348-62页
[0037]
[非专利文献6]《自然生物技术》,第26卷(第3期):313-315页,2008年
[0038]
[非专利文献7]《美国国家科学院院刊》,第111卷:12426-12431页,2014年
[0039]
[非专利文献8]《新英格兰医学杂志》,第346卷:1185-1193页,2002年
[0040]
[非专利文献9]《自然》,第302卷:415-419页,2003年
技术实现要素:
[0041]
[技术问题]
[0042]
通过将诸如基因之类的诱导物转移到细胞中而建立诱导干细胞,诸如ips细胞。然后,对这些细胞进行扩增培养,并冷冻保存。然而,临床ips细胞(例如,glp或gmp等级)的生产和工业化存在以下问题:
[0043]
1)成本
[0044]
临床ips细胞需要生产并保存在完全清洁无菌的“洁净室”中。然而,保持所需的清洁度是非常昂贵的。因此,ips细胞的生产是昂贵的,这对工业化是一个重大的障碍。
[0045]
2)质量
[0046]
从建立干细胞到保存干细胞的过程是复杂的,并且需要许多人工技术。此外,干细胞的生产部分地取决于操作者的技能。因此,取决于生产者或实验批次,ips细胞的质量可有所不同。
[0047]
3)时间
[0048]
为了防止与属于特定供体以外的个体的ips细胞的交叉污染,在洁净室内任何给定时间段内仅生产来自单人的ips细胞。另外,ips细胞的建立和质量评估都需要很长时间。因为在每个室内一次仅对一个个体生产ips细胞,所以对许多个体生产ips细胞需要很长时间。
[0049]
4)人力资源
[0050]
如上文所述及,目前,ips细胞的生产在很大程度上取决于人工过程。同时,只有少数技术人员具备生产临床ips细胞的必要技能。
[0051]
从建立干细胞到其保存的一系列程序较复杂,这种复杂性是不利的。响应于此,本发明的目的是提供一种干细胞制造系统,该系统使得有可能制造干细胞。
[0052]
(关于诱导干细胞的方法、用于干细胞的漂浮培养方法和用于干细胞的漂浮培养容器的目的)
[0053]
ips细胞在贴壁培养系统中的培养需要培养皿,并且因此需要非常大的空间,从而导致培养效率差。在诱导ips细胞之后或其扩增培养期间,ips细胞必须从培养皿中分离。然而,从培养皿中分离ips细胞的过程大大地损伤了ips细胞。此外,这些程序复杂,并且不适合机械化。
[0054]
在制备源自小鼠的饲养细胞、在培养皿中在饲养细胞层上生产和扩增培养ips细胞的情况下,ips细胞被源自动物的成分污染。因此,与饲养细胞共培养的ips细胞不适于临床使用。或者,在没有饲养细胞的情况(无饲养细胞的情况)下生产和扩增培养ips细胞给ips细胞造成压力。这种压力使得ips细胞可能发展出核型异常或染色体损伤。此外,当不使用饲养细胞时,必须对培养皿施加特殊的涂层,这使得程序更加复杂。
[0055]
在贴壁培养系统中培养ips细胞的情况下,ips细胞仅可二维增殖,并且因此不利地表现出较差的生长效率。
[0056]
相比之下,可能的是在三维培养(漂浮培养)系统中培养ips细胞。然而,在常规的漂浮培养系统中,必须连续搅拌培养液以防止ips细胞下沉。然而,当搅拌培养液时,ips细胞相互碰撞,并从而损伤。这不利地导致细胞死亡或核型异常。
[0057]
在常规的漂浮培养系统中,ips细胞随机聚集并相互结合以形成各种大小的细胞簇(集落)。因此,集落之间不可保持均匀的大小分布。如果集落变得太大,那么营养物质或生长因子不能扩散到集落中心的细胞,这导致这些最内层细胞的分化或细胞死亡。相反,如果集落太小,那么它们不适合继代培养。
[0058]
ips细胞来源于单个体细胞。因此,每个ips细胞系在很小程度上可具有不同的性质。因此,独立培养每个集落并建立单独的ips细胞系是非常重要的。在这方面,当在漂浮培养系统中培养ips细胞时,需要确保ips细胞的集落独立生长并彼此分离。
[0059]
在贴壁培养系统中,各自源自单一体细胞的ips细胞独立形成集落。然而,如上文所述及,在常规的漂浮培养系统中,ips细胞彼此随机聚集以形成集落。因此,在常规的漂浮系统中生产的集落不可保持克隆性。因此,尚未有经由常规的漂浮培养系统诱导和培养ips细胞的尝试成功地生产源自个体细胞的ips集落。相应地,尚未开发出使得有可能建立独立的ips细胞系的常规的漂浮培养方法。
[0060]
因此,本发明的另一个目的是提供一种诱导干细胞的方法、一种用于干细胞的漂浮培养方法和一种用于干细胞的漂浮培养容器,其使得有可能培养具有隔离且分离的集落的ips细胞。
[0061]
(关于生产诱导多能干细胞的方法的目的)
[0062]
本发明的另一个目的是提供一种生产临床可用的干细胞的方法。
[0063]
(关于从动物细胞生产特定体细胞的方法的目的)
[0064]
本发明的另一个目的是提供一种在短时间段内并且在不引起基因损伤的情况下从另一种类型的动物细胞高效地生产特定类型的体细胞的方法。
[0065]
[问题的解决方案]
[0066]
本发明的一个方面提供了一种干细胞制造系统,包括:(a)转移前细胞溶液发送通道,含有细胞的溶液流动通过该转移前细胞溶液发送通道;(b)诱导物溶液发送机构,其将多能性诱导物送入转移前细胞溶液发送通道;(c)诱导物转移装置,其连接到转移前细胞溶液发送通道并将多能性诱导物转移到细胞中以生产携带诱导物的细胞;(d)细胞簇生产装置,其培养携带诱导物的细胞以生产多个由干细胞组成的细胞簇;(e)包装装置,其依次包装多个细胞簇;以及(f)容器,其容纳转移前细胞溶液发送通道、诱导物溶液发送机构、诱导物转移装置、细胞簇生产装置和包装装置。
[0067]
上述干细胞制造系统还可包括分离装置,其从血液中分离细胞,其中含有由分离装置分离的细胞的溶液可流动通过转移前细胞溶液发送通道。
[0068]
在上述干细胞制造系统中,细胞簇生产装置可包括:重编程培养装置,其培养由诱导物转移装置生产的携带诱导物的细胞;第一分割机构,其将由重编程培养装置所建立的干细胞组成的细胞簇分割成多个细胞簇;扩增培养装置,其扩增培养由第一分割机构分割的多个细胞簇;第二分割机构,其将由扩增培养装置所扩增培养的干细胞组成的细胞簇分
割成多个细胞簇;以及细胞簇输送机构,其将多个细胞簇依次送入包装装置。
[0069]
重编程培养装置可以包括第一培养液补给装置和扩增培养装置,该第一培养液补给装置用培养液补给携带诱导物的细胞,该扩增培养装置可包括第二培养液补给装置,该第二培养液补给装置用培养液补给多个细胞簇。
[0070]
上述干细胞制造系统还可包括:重编程培养摄影装置,其拍摄由重编程培养装置培养的细胞;以及扩增培养摄影装置,其拍摄由扩增培养装置培养的细胞,其中在重编程培养装置和扩增培养装置中可使用无色培养液。
[0071]
在上述干细胞制造系统中,转移前细胞溶液发送通道的内壁可对细胞无粘附性。
[0072]
在上述干细胞制造系统中,转移前细胞溶液发送通道和诱导物溶液发送机构可设置在基底上。
[0073]
在上述干细胞制造系统中,包装装置可使用peltier设备或液氮来冷冻细胞簇。或者,包装装置可以通过诸如蒸气压缩或蒸气吸收的冷冻方法冷冻细胞簇。
[0074]
上述干细胞制造系统还可包括空气净化装置,其净化容器内的气体。
[0075]
上述干细胞制造系统还可包括温度控制装置,其控制容器内的气体的温度。
[0076]
上述干细胞制造系统还可包括二氧化碳浓度控制装置,其控制容器内的气体的二氧化碳浓度。
[0077]
上述干细胞制造系统还可包括灭菌装置,其对容器的内部进行干热灭菌或气体灭菌。
[0078]
在上述干细胞制造系统中,诱导物溶液发送机构、诱导物转移装置、细胞簇生产装置和包装装置可由服务器基于操作程序进行调控,并且服务器可监控是否基于操作程序来操作诱导物溶液发送机构、诱导物转移装置、细胞簇生产装置和包装装置,并作操作记录。
[0079]
上述干细胞制造系统还可包括将诱导物转移到干细胞中以使干细胞分化成体细胞的装置。
[0080]
本发明的一个方面提供了一种诱导干细胞的方法,包括从在凝胶培养基中漂浮培养的体细胞诱导干细胞。
[0081]
在上述诱导干细胞的方法中,可不搅拌凝胶培养基。凝胶培养基可为用脱乙酰结冷胶凝胶化的培养基。
[0082]
在上述诱导干细胞的方法中,凝胶培养基可不含生长因子。或者,凝胶培养基可含有浓度为40重量%或更低的生长因子。
[0083]
在上述诱导干细胞的方法中,凝胶培养基可不含bfgf。凝胶培养基可包括人es/ips培养基。
[0084]
本发明的一个方面还提供了一种用于干细胞的漂浮培养方法,包括在不含生长因子的凝胶培养基中漂浮培养干细胞。
[0085]
本发明的一个方面还提供了一种用于干细胞的漂浮培养方法,包括在含有浓度为40重量%或更低的生长因子的凝胶培养基中漂浮培养干细胞。
[0086]
本发明的一个方面还提供了一种用于干细胞的漂浮培养方法,包括在不含bfgf的凝胶培养基中漂浮培养干细胞。
[0087]
本发明的一个方面还提供了一种用于干细胞的漂浮培养方法,包括在含有浓度为400μg/l或更低的bfgf的凝胶培养基中漂浮培养干细胞。
[0088]
在上述用于干细胞的漂浮培养方法中,可不搅拌凝胶培养基。凝胶培养基可为用脱乙酰结冷胶凝胶化的培养基。凝胶培养基可含有rock抑制剂。干细胞在凝胶培养基中的浓度可为0.1
×
105个细胞/ml或更高。
[0089]
上述用于干细胞的漂浮培养方法还可包括在漂浮培养之前将干细胞解离成单细胞,并将解离成单细胞的干细胞置于凝胶培养基中。
[0090]
在上述用于干细胞的漂浮培养方法的漂浮培养中,单细胞可在保持其克隆性的同时形成集落。
[0091]
上述用于干细胞的漂浮培养方法还可包括在漂浮培养之前,使用格栅板悬滴培养干细胞以形成集落,并将形成集落置于凝胶培养基中。
[0092]
在上述用于干细胞的漂浮培养方法中,干细胞可在保持其未分化状态的同时进行增殖。
[0093]
本发明的一个方面还提供一种用于干细胞的漂浮培养容器,包括:透析管,其容纳干细胞和凝胶培养基;容器,其容纳透析管,其中凝胶培养基置于透析管周围。
[0094]
在上述用于干细胞的漂浮培养容器中,透析管的截留分子量可为0.1kda或更大。透析管可由选自纤维素酯、纤维素酯衍生物、再生纤维素和醋酸纤维素中的至少一种制成。
[0095]
本发明的一个方面还提供了一种用于干细胞的漂浮培养方法,包括:将干细胞和凝胶培养基置于透析管中;将透析管置于容器中;将凝胶培养基置于容器中透析管周围;以及在透析管中的凝胶培养基中漂浮培养干细胞。将干细胞和凝胶培养基置于透析管中,将透析管置于容器中,和将凝胶培养基置于容器中透析管周围的顺序未特别地限制。例如,可将透析管置于容器中,然后可将干细胞和凝胶培养基置于透析管中。
[0096]
在上述用于干细胞的漂浮培养方法中,透析管的截留分子量可以为0.1kda或更大。透析管可由选自纤维素酯、纤维素酯衍生物、再生纤维素和醋酸纤维素中的至少一种制成。
[0097]
在上述用于干细胞的漂浮培养方法中,透析管周围的凝胶培养基中可补充有rock抑制剂。可不搅拌凝胶培养基。凝胶培养基可为用脱乙酰结冷胶凝胶化的培养基。
[0098]
在上述用于干细胞的漂浮培养方法中,凝胶培养基可不含生长因子。或者,凝胶培养基可含有浓度为40重量%或更低的生长因子。
[0099]
在上述用于干细胞的漂浮培养方法中,凝胶培养基可不含bfgf。
[0100]
在上述用于干细胞的漂浮培养方法中,凝胶培养基中的干细胞的浓度可为0.1
×
105个细胞/ml或更高。
[0101]
上述用于干细胞的漂浮培养方法还可以包括在漂浮培养之前,将干细胞解离成单细胞,并将解离成单细胞的干细胞置于凝胶培养基中。
[0102]
在上述用于干细胞的漂浮培养方法的漂浮培养中,单细胞可在保持其克隆性的同时形成集落。
[0103]
上述用于干细胞的漂浮培养方法还可以包括在漂浮培养之前,使用格栅板悬滴培养干细胞以形成集落,并将形成集落置于凝胶培养基中。
[0104]
在上述用于干细胞的漂浮培养方法中,干细胞可在保持其未分化状态的同时进行增殖。
[0105]
上述用于干细胞的漂浮培养方法还可包括用新鲜凝胶培养基更换容器中透析管
周围的凝胶培养基。
[0106]
上述用于干细胞的漂浮培养方法还可包括用新鲜凝胶培养基补充容器中透析管周围的凝胶培养基。
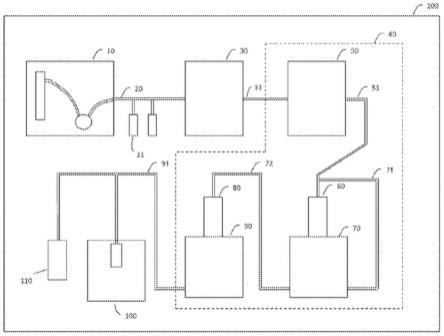
[0107]
在上述用于干细胞的漂浮培养方法中,可不更换透析管中的凝胶培养基。凝胶培养基可包括人es/ips培养基。
[0108]
本发明的一个方面还提供了一种通过漂浮诱导干细胞的方法,包括:将体细胞和凝胶培养基置于透析管中;将透析管置于容器中;将凝胶培养基置于容器中透析管周围;以及从漂浮在透析管中的凝胶培养基中的体细胞诱导干细胞。将体细胞和凝胶培养基置于透析管中,将透析管置于容器中,和将凝胶培养基置于容器中透析管周围的顺序未特别地限制。例如,可将透析管置于容器中,然后可将体细胞和凝胶培养基置于透析管中。
[0109]
在上述通过漂浮诱导干细胞的方法中,透析管的截留分子量可为0.1kda或更大。透析管可由选自纤维素酯、纤维素酯衍生物、再生纤维素和醋酸纤维素中的至少一种制成。
[0110]
在上述通过漂浮诱导干细胞的方法中,可不搅拌凝胶培养基。凝胶培养基可为用脱乙酰结冷胶凝胶化的培养基。
[0111]
在上述通过漂浮诱导干细胞的方法中,凝胶培养基可不含生长因子。
[0112]
在上述通过漂浮诱导干细胞的方法中,凝胶培养基可不含bfgf。
[0113]
上述通过漂浮诱导干细胞的方法还可以包括在漂浮培养之前,将体细胞解离成单细胞,并将解离成单细胞的体细胞置于凝胶培养基中。
[0114]
在上述通过漂浮诱导干细胞的方法的漂浮培养中,单细胞可在保持其克隆性的同时形成集落。
[0115]
上述通过漂浮诱导干细胞的方法还可包括用新鲜凝胶培养基更换容器中透析管周围的凝胶培养基。
[0116]
上述通过漂浮诱导干细胞的方法还可包括用新鲜凝胶培养基补充容器中透析管周围的凝胶培养基。
[0117]
在上述通过漂浮诱导干细胞的方法中,可不更换透析管中的凝胶培养基。凝胶培养基可包括人es/ips培养基。
[0118]
本发明的一个方面还提供了一种用于生产诱导多能干细胞的方法,其包括:制备体细胞;以及通过脂质转染法将重编程因子rna转移到体细胞中。
[0119]
在上述生产诱导多能干细胞的方法中,体细胞可为血细胞。血细胞可为单核细胞。血细胞可为造血干/祖细胞。血细胞可呈cd34阳性。血细胞可为在细胞呈cd34阳性的条件下分离的血细胞。血细胞可呈cd3阳性。血细胞可在细胞呈cd3阳性的条件下分离。
[0120]
在上述生产诱导多能干细胞的方法中,重编程因子rna可包括oct3/4mrna、sox2 mrna、klf4 mrna和c-myc mrna。重编程因子rna还可包括选自由glis1 mrna、foxh1 mrna、l-myc mrna和p53-dn mrna组成的组中的至少一种。重编程因子rna还可包括lin28a mrna或lin28bmrna。
[0121]
在上述生产诱导多能干细胞的方法中,sirna脂质转染试剂或mrna脂质转染试剂可用于以重编程因子rna的脂质转染中。
[0122]
在上述生产诱导多能干细胞的方法中,选自lipofectamine(r)rnaimax转染试剂、lipofectamine(r)messengermax转染试剂、stemfect(r)rna转染试剂和reprorna(r)转染
试剂中的至少一种可用于以重编程因子rna的脂质转染中。
[0123]
在上述生产诱导多能干细胞的方法中,用于以重编程因子rna的脂质转染的血细胞的数量可为1至1
×
108个细胞。用于以重编程因子rna的脂质转染的重编程因子rna的量可为每次5ng至50μg。用于以重编程因子rna的脂质转染的脂质转染试剂的量可为每次0.1μl至500μl。以重编程因子rna的脂质转染可每次进行0.1小时或更长和24小时或更短。以重编程因子rna的脂质转染可以进行多次。
[0124]
在上述生产诱导多能干细胞的方法中,用于以重编程因子rna的脂质转染的培养基可为opti-mem(r)。
[0125]
上述生产诱导多能干细胞的方法还可包括使用过滤器从血液中分离单核细胞。
[0126]
本发明的一个方面还提供了一种从动物细胞生产特定体细胞的方法,包括:制备动物细胞;以及通过脂质转染将诱导物rna转移到动物细胞中,以将动物细胞分化成体细胞。
[0127]
在上述从动物细胞生产特定体细胞的方法中,动物细胞可为干细胞。干细胞可为诱导多能干细胞。干细胞可为ips细胞。干细胞可为胚胎干细胞。
[0128]
在上述从动物细胞生产特定体细胞的方法中,动物细胞可为人成纤维细胞。或者,动物细胞可为血细胞。
[0129]
在上述从动物细胞生产特定体细胞的方法中,诱导物rna可包括对应于药物抗性基因的mrna。
[0130]
上述从动物细胞生产特定体细胞的方法还可包括选择在脂质转染之后表现出药物抗性的细胞。
[0131]
上述从动物细胞生产特定体细胞的方法,诱导物rna可包括对应于嘌呤霉素抗性基因的mrna。
[0132]
上述从动物细胞生产特定体细胞的方法还可包括选择在脂质转染之后表现出嘌呤霉素抗性的细胞。
[0133]
在上述从动物细胞生产特定体细胞的方法中,体细胞可为神经元细胞。诱导物rna可包括ngn2 mrna。诱导神经元细胞可呈ngn2阳性。诱导神经元细胞可呈β-iii tubulin、map2、psa-ncam或vglut阳性。
[0134]
在上述从动物细胞生产特定体细胞的方法中,messengermax(r)可用于以诱导物rna的脂质转染中。
[0135]
在上述从动物细胞生产特定体细胞的方法中,用于以诱导物rna的脂质转染的细胞的数量可为1
×
104至1
×
108个细胞。用于以诱导物rna的脂质转染的诱导物rna的量可为每次200ng至5000ng。用于以诱导物rna的脂质转染的脂质转染试剂的量可为每次0.1μl至100μl。
[0136]
在上述从动物细胞生产特定体细胞的方法中,用于以诱导物rna的脂质转染的培养基可为opti-mem(r)。
[0137]
在上述从动物细胞生产特定体细胞的方法中,动物细胞可在从以诱导物rna的脂质转染的10天内分化成体细胞。
[0138]
在上述从动物细胞生产特定体细胞的方法中,通过脂质转染将诱导物rna转移到动物细胞中可重复多次。
[0139]
在上述从动物细胞生产特定体细胞的方法中,可在涂覆有基膜基质的基底上培养动物细胞。
[0140]
在上述从动物细胞生产特定体细胞的方法中,可在含有b18r的培养基中培养动物细胞。或者,可在不含b18r的培养基中培养动物细胞。
[0141]
[发明的有益效果]
[0142]
本发明使得有可能提供能够制造干细胞的干细胞制造系统。
[0143]
(诱导干细胞的方法、用于干细胞的漂浮培养方法和用于干细胞的漂浮培养容器的有益效果)
[0144]
本发明使得有可能提供一种诱导干细胞的方法、一种用于干细胞的漂浮培养方法,以及一种用于干细胞的漂浮培养容器,其能够在ips细胞的集落分开的情况下培养ips细胞。
[0145]
(生产诱导多能干细胞的方法的有益效果)
[0146]
本发明使得有可能提供一种用于生产临床可用的诱导多能干细胞的方法。
[0147]
(从动物细胞生产特定体细胞的方法的有益效果)
[0148]
本发明使得有可能提供一种从动物细胞生产特定体细胞的方法,其能够在短时间内高效生产特定体细胞而不损伤动物细胞的基因。
附图说明
[0149]
[图1]图1是根据本发明实施例的干细胞制造系统的示意图。
[0150]
[图2]图2是根据本发明实施例的干细胞制造系统中的转移后细胞溶液发送通道的一个实例的示意性横截面图。
[0151]
[图3]图3是根据本发明实施例的干细胞制造系统中的转移后细胞溶液发送通道的一个实例的示意性横截面图。
[0152]
[图4]图4是根据本发明实施例的干细胞制造系统中使用的培养袋的示意图。
[0153]
[图5]图5是示出根据本发明的第二实施例的用于干细胞的漂浮培养容器的示意图。
[0154]
[图6]图6是根据实例1的ips细胞的集落的照片。
[0155]
[图7]图7是根据实例1的ips细胞的集落的照片。
[0156]
[图8]图8是根据实例1的ips细胞的集落的照片。
[0157]
[图9]图9是示出根据实例1的ips细胞的集落的分化状态的图。
[0158]
[图10]图10是根据实例2的ips细胞的集落的照片。
[0159]
[图11]图11是根据实例3的ips细胞的集落的照片。
[0160]
[图12]图12是根据实例3的ips细胞的集落的照片。
[0161]
[图13]图13是根据实例4的ips细胞的照片。
[0162]
[图14]图14是示出根据实例4的ips细胞的集落的数量的图。
[0163]
[图15]图15是根据实例4的ips细胞的集落的照片。
[0164]
[图16]图16是根据实例5的ips细胞的集落的照片。
[0165]
[图17]图17是示出根据实例5的ips细胞的每种密度的集落形成速率的图。
[0166]
[图18]图18是示出根据实例5的每种培养基量的集落形成率的图。
[0167]
[图19]图19是根据实例6的ips细胞的照片。
[0168]
[图20]图20是示出根据实例6的ips细胞的集落的数量的图。
[0169]
[图21]图21是根据实例7的ips细胞的集落的照片。
[0170]
[图22]图22是示出根据实例7的各种培养条件下的ips细胞的集落的数量的图。
[0171]
[图23]图23是根据实例7的每种培养基的ips细胞的集落的照片。
[0172]
[图24]图24是示出根据实例7的ips细胞的集落的分化状态的图。
[0173]
[图25]图25是根据实例8的ips细胞的照片。
[0174]
[图26]图26是示出根据实例8的每种培养基量的集落形成率的图。
[0175]
[图27]图27是根据实例9的凝胶培养基的照片。
[0176]
[图28]图28是根据实例9的ips细胞的集落的照片。
[0177]
[图29]图29是根据实例10的ips细胞的集落的照片。
[0178]
[图30]图30是根据实例11的ips细胞的集落的照片。
[0179]
[图31]图31是根据实例12的ips细胞的集落的照片。
[0180]
[图32]图32是示出根据实例12的ips细胞的集落的大小的图。
[0181]
[图33]图33是根据实例12的ips细胞的集落的照片。
[0182]
[图34]图34是示出根据实例12的ips细胞的集落的分化状态的图。
[0183]
[图35]图35是根据实例13的荧光显微镜照片。
[0184]
[图36]图36是示出根据实例13的使用荧光激活的流式细胞仪的分析结果的图。
[0185]
[图37]图37是根据实例14的细胞的照片。
[0186]
[图38]图38是根据实例14的细胞的照片。
[0187]
[图39]图39是示出根据实例14的转染效率和存活率的百分比的图。
[0188]
[图40]图40是根据实例15的细胞的照片。
[0189]
[图41]图41是根据实例15的在荧光显微镜下观察细胞拍摄的照片。
[0190]
[图42]图42是示出根据实例15的tuj-1阳性细胞的百分比的图。
[0191]
[图43]图43示出了根据实例15的细胞的照片。
[0192]
[图44]图44是根据实例16的转染方法的示意图。
[0193]
[图45]图45是根据实例16的细胞的照片。
[0194]
[图46]图46示出了根据实例16的细胞的照片。
具体实施方式
[0195]
在下文中,将描述本发明的实施例。在附图的下述描述中,相同或相似的附图标记将用于指代相同或相似的部分。然而,附图是示意性的。因此,应根据下面的描述判断具体的尺寸等。另外,应理解,附图之间尺寸的关系或比率可不同。
[0196]
(第一实施例)
[0197]
如图1所示,根据本发明第一实施例的干细胞制造系统包括:分离装置10,其从血液中分离细胞;转移前细胞溶液发送通道20,含有由分离装置10分离的细胞的溶液流动通过转移前细胞溶液发送通道20;诱导物溶液发送机构21,其将多能性诱导物送入转移前细胞溶液发送通道20;诱导物转移装置30,其连接到转移前细胞溶液传输通道20并将多能性诱导物转移到细胞中以生产携带诱导物的细胞;细胞簇生产装置40,其培养携带诱导物的
细胞以生产多个由干细胞组成的细胞簇;以及包装装置100,其依次包装多个细胞簇。
[0198]
干细胞制造系统还包括容器200,其容纳分离装置10、转移前细胞溶液发送通道20、诱导物溶液发送机构21、诱导物转移装置30、细胞簇生产装置40和包装装置100。
[0199]
干细胞制造系统还可包括:空气净化装置,其净化容器200内的气体:温度控制装置,其控制容器200内的气体的温度;以及二氧化碳浓度控制装置,其控制容器200内的气体的二氧化碳(co2)浓度。空气净化装置可包括清洁度传感器,其监控容器200内的气体的清洁度。空气净化装置使用例如hepa(高效微粒空气)过滤器来净化容器200中的空气。根据例如iso标准14644-1,空气净化装置将容器200中的空气的清洁度保持在iso 1和iso 6之间的清洁度。温度控制装置可包括监控容器200内的气体的温度的温度传感器。co2浓度控制装置可包括监控容器200内的气体的co2浓度的co2浓度传感器。
[0200]
容器200设置有例如门。在其中门关闭的状态下,内部被完全密封,使得内部空气的清洁度、温度和co2浓度可保持恒定。容器200优选是透明的,使得可从外部观察装置的内部状态。容器200可为一体地包括诸如橡胶手套之类的手套的手套箱。
[0201]
分离装置10接收例如含有人血液的小瓶。分离装置10包括例如储存诸如乙二胺四乙酸(edta)、肝素和柠檬酸葡萄糖配方a溶液(acd-a溶液,terumo股份有限公司)的抗凝剂的抗凝剂槽。分离装置10使用泵等将来自抗凝剂槽的抗凝剂添加至人血液。
[0202]
分离装置10包括例如分离用试剂槽,其储存单核细胞分离用试剂,诸如ficoll-paque premium(ge healthcare japan股份有限公司)。分离装置10使用泵等从分离用试剂槽以5ml/管将单核细胞分离用试剂分配到例如2个15ml管。注意,可使用树脂袋代替管。
[0203]
分离装置10还包括储存诸如磷酸盐缓冲盐水(pbs)的缓冲溶液的缓冲溶液槽。分离装置10通过使用泵等添加得自缓冲液槽的5ml的缓冲液来稀释例如5ml的人血液。此外,分离装置10使用泵等将5ml的稀释后的人血液添加至各管中的单核细胞分离用试剂。
[0204]
分离装置10还包括其中可设定温度的离心机。离心机温度设定为例如18℃。分离装置10使用运输装置等将含有单核细胞分离用试剂和人血液等的各管置于离心机的支架中。离心机将管中的溶液以例如400
×
g离心30分钟。代替管,可对树脂袋进行离心。
[0205]
在离心之后,分离装置10使用泵等回收管内溶液中由单核细胞组成的白浊的中间层。分离装置10使用泵等将回收的单核细胞悬浮液送入转移前细胞溶液发送通道20。或者,分离装置10还向例如2ml的回收单核细胞溶液添加例如12ml的pbs,并将管置于离心机的支架中。离心机将管中的溶液以例如200
×
g离心10分钟。
[0206]
在离心之后,分离装置10使用泵等吸移管内溶液的上清液,并通过添加3ml的诸如x-vivo10(r)(lonza japan有限责任公司)的单核细胞培养基,使单核细胞溶液悬浮于管中。分离装置10使用泵等将单核细胞悬浮液送入转移前细胞溶液发送通道20。分离装置10可使用透析膜从血液中分离单核细胞。或者,在使用预先从皮肤等分离的诸如成纤维细胞的体细胞的情况下,分离装置10可为不必要的。
[0207]
分离装置10可以通过离心分离以外的方法分离适于诱导的细胞。当待分离的细胞为例如t细胞时,可通过淘选来分离对于cd3、cd4和cd8中的任一者呈阳性的细胞。当待分离的细胞是血管内皮祖细胞时,可通过淘选分离对于cd34呈阳性的细胞。当待分离的细胞是b细胞时,可通过淘选来分离对于cd10、cd19和cd20中的任一者呈阳性的细胞。分离方法不限于淘选,并且可通过磁性细胞分离方法、流式细胞术或其他方法分离细胞。或者,分离装置
10可通过后文提及的实施例中描述的方法分离适于诱导的细胞。例如,如第五实施例中所述,可基于细胞表面标记使用磁分离装置来分离适于诱导的细胞。或者,可使用过滤器来分离适于诱导的细胞。待诱导的细胞不限于血细胞,可为成纤维细胞等。
[0208]
诱导物溶液发送机构21包括储存诱导物转移试剂溶液等的诱导物转移试剂槽。诸如基因转移试剂溶液的诱导物转移试剂溶液含有例如诸如人t细胞nucleofector(r)(lonza japan有限责任公司)溶液的电穿孔溶液、补充溶液和质粒组。质粒组含有例如0.83μg的pcxle-hoct3/4-shp53-f、0.83μg的pcxle-hsk、0.83μg的pce-hul和0.5μg的pcxwb-ebna1。或者,诱导物转移试剂溶液可含有后文提及的的第四和第五实施例中描述的试剂等。例如,如第五实施例中所述,可通过脂质转染法将编码重编程因子的rna转移到细胞中。诱导物溶液发送机构21使用微型泵等将诱导物转移试剂溶液送入转移前细胞溶液发送通道20,使得单核细胞悬液悬浮在诱导物转移试剂溶液中。
[0209]
通过用聚-hema(聚-2-羟乙基甲基丙烯酸酯)涂覆,转移前细胞溶液发送通道20的内壁可对细胞无粘附性,以防止细胞粘附到内壁上。或者,抗细胞粘附的材料可用作转移前细胞溶液发送通道20的材料。另外,具有高测温传导率的co2可渗透材料可用作转移前细胞溶液发送通道20的材料,使得转移前细胞溶液发送通道20的内部条件等同于容器200内的受控温度和co2浓度。根据防止污染的观点,转移前细胞溶液发送通道20还可设置有逆流防止阀。
[0210]
连接到转移前细胞溶液发送通道20的诱导物转移装置30是例如电穿孔机,其接收诱导物转移试剂溶液和单核细胞悬浮液的混合溶液并用质粒进行单核细胞的电穿孔。在电穿孔之后,诱导物转移装置30将单核细胞培养基添加至含有用质粒电穿孔的单核细胞的溶液中。诱导物转移装置30使用泵等将含有用质粒电穿孔的单核细胞(以下称为“携带诱导物的细胞”)的溶液送入转移后细胞溶液发送通道31。注意,诱导物转移装置30不限于电穿孔机。诱导物转移装置30可通过后文提及的第四和第五实施例中描述的方法将诱导物转移到细胞中。培养基可为凝胶培养基。在这种情况下,凝胶培养基可例如不含诸如碱性成纤维细胞生长因子(bfgf)的生长因子。或者,凝胶培养基可含有400μg/l或更低、40μg/l或更低或10μg/l或更低的低浓度的诸如bfgf的生长因子。凝胶培养基可不含tgf-β,或可含有600ng/l或更低、300ng/l或更低或100ng/l或更低的低浓度的tgf-β。
[0211]
转移后细胞溶液发送通道31的内壁可通过涂覆聚-hema保持为非粘附性的,以防止细胞粘附于内壁上。或者,抗细胞粘附的材料可用作转移后细胞溶液发送通道31的材料。另外,具有高测温传导率的co2可渗透材料可用作转移后细胞溶液发送通道31的材料,使得转移后细胞溶液发送通道31的内部条件等同于容器200内的受控温度和co2浓度。根据防止污染的观点,转移后细胞溶液发送通道31还可设置有逆流防止阀。在电穿孔之后,许多细胞死亡,并且死细胞可能形成细胞簇。因此,转移后细胞溶液发送通道31可设置有除去死细胞簇的过滤器。或者,如图2所示,可在转移后细胞溶液发送通道31的内部设置间歇地改变内径的一个或多个壁。或者,如图3所示,可间歇地改变转移后细胞溶液发送通道31的内径。
[0212]
连接到转移后细胞溶液发送通道31的细胞簇生产装置40包括:重编程培养装置50,其培养携带由诱导物转移装置30生产的诱导物的细胞;第一分割机构60,其将由重编程培养装置50所建立的干细胞组成的细胞簇分割成多个细胞簇;扩增培养装置70,其扩增培养由第一分割机构60分割的多个细胞簇;第二分割机构80,其将由扩增培养装置70所扩增
培养的干细胞组成的细胞簇分割成多个细胞簇;以及细胞簇输送机构90,其将多个细胞簇依次送入包装装置100。
[0213]
重编程培养装置50可在其中容纳孔板。重编程培养装置50还包括移液机。重编程培养装置50从转移后细胞溶液发送通道31接收含有携带诱导物的细胞的溶液,并通过移液机将溶液分配到孔。重编程培养装置50在将携带诱导物的细胞分配到孔例如三天、五天和七天之后,添加干细胞培养基,诸如stemfit(r)(ajinomoto股份有限公司)。可添加碱性成纤维细胞生长因子(碱性fgf)作为培养基的补充。注意,可向培养基添加连续供给fgf-2(碱性fgf、bfgf或fgf-b)的持续释放珠,诸如stem beads fgf2(funakoshi有限责任公司)。因为fgf有时不稳定,所以可通过将肝素模拟聚合物偶联至fgf来稳定fgf。重编程培养装置50还更换培养基,例如,在将携带诱导物的细胞分配到孔中9天之后,并且以后每两天更换培养基,直到ips细胞的细胞簇(集落)超过1mm。
[0214]
在形成细胞簇之后,重编程培养装置50通过移液机回收细胞簇,并向回收的细胞簇添加作为胰蛋白酶的替代物的重组酶,诸如tryple select(r)(life technologies股份有限公司)。重编程培养装置50还将含有回收的细胞簇的容器置于培养箱中,在培养箱中细胞簇与作为胰蛋白酶的替代物的重组酶在37℃下在5%co2环境中反应10分钟。或者,重编程培养装置50可通过使用移液机移液来破坏细胞簇。作为另一种替代方案,重编程培养装置50可通过使细胞簇穿过设有过滤器的管或内径间歇地变化的管来破坏细胞簇,如同如图2或图3所示的转移后细胞溶液发送通道31。然后,重编程培养装置50向含有破裂的细胞簇的溶液添加用于多能干细胞的培养基,诸如stemfit(r)(ajinomoto股份有限公司)。
[0215]
重编程培养装置50中的培养可以在co2可渗透袋中而非孔板中进行。培养可为贴壁培养,或可为漂浮培养。在漂浮培养的情况下,可对培养进行搅拌。培养基可为琼脂形式。琼脂形式的培养基的示例包括结冷胶聚合物。当使用琼脂形式的培养基时,即使以漂浮培养的形式,不需要搅拌并且有可能生产源自一个细胞的单细胞簇,因为细胞既不下沉也不粘附。重编程培养装置50中的培养可为悬滴培养。
[0216]
重编程培养装置50可包括第一培养液补给装置,其对孔板或co2可渗透袋补给培养液。第一培养液补给装置可回收孔板或co2可渗透袋中的培养液,使用过滤器或透析膜过滤培养液,并循环纯化后的培养液。在这种情况下,可向待循环的培养液中添加生长因子等。重编程培养装置50还可包括例如控制培养液温度的温度控制装置和控制培养液附近湿度的湿度控制装置。
[0217]
在重编程培养装置50中,例如,如图4所示,可将细胞置于诸如透析膜的培养液可渗透袋301中,并且可将培养液可渗透袋301置于培养液不可渗透且co2可渗透的袋302中,而培养液可置于袋301和302中。可制备多个包含新鲜培养液的袋302,并且重编程培养装置50可以预定的时间间隔用另一包含新鲜培养液的袋302替换其中置入有包含细胞的袋301的袋302。注意,重编程培养装置50中的培养方法不限于上述的方法,并且培养可以通过后文提及的第二和第三实施例中描述的方法来进行。例如,如第二实施例中所述,可使用凝胶培养基。在这种情况下,凝胶培养基可例如不含诸如碱性成纤维细胞生长因子(bfgf)的生长因子。或者,凝胶培养基可含有400μg/l或更低、40μg/l或更低或10μg/l或更低的低浓度的诸如bfgf的生长因子。凝胶培养基可不含tgf-β,或者可含有600ng/l或更低、300ng/l或更低或100ng/l或更低的低浓度的tgf-β。如第三实施例中所述,可使用一种漂浮培养容器,
包括:容纳干细胞和凝胶培养基的透析管;以及容纳透析管的容器,其中凝胶培养基置于透析管周围。
[0218]
干细胞制造系统还可包括拍摄重编程培养装置50中的培养的重编程培养摄影装置。注意,当将无色培养液用作用于重编程培养装置50的培养液时,有可能抑制使用有色培养液时可发生的漫反射或自发荧光。因为诱导细胞和未诱导细胞在细胞形状和大小等方面不同,所以干细胞制造系统还可包括诱导状态监控装置,其通过拍摄重编程培养装置50中的细胞计算诱导细胞的百分比。或者,诱导状态监控装置可通过抗体免疫染色方法或rna提取方法鉴定诱导细胞的百分比。干细胞制造系统还可包括未诱导细胞除去装置,其通过磁性细胞分离方法、流式细胞术等除去未诱导细胞。
[0219]
第一细胞簇溶液发送通道51连接到重编程培养装置50。重编程培养装置50使用泵等将含有作为胰蛋白酶的替代物的重组酶和细胞簇的溶液送入第一细胞簇溶液发送通道51。当细胞簇可以被物理破坏时,作为胰蛋白酶的替代物的重组酶可为不必要的。第一细胞簇溶液发送通道51可连接到分支通道,该分支通道具有仅允许具有小于预定大小的诱导细胞穿过并除去具有预定大小或更大的未诱导细胞的内径。
[0220]
第一细胞簇溶液发送通道51的内壁可通过涂覆聚hema而对细胞无粘附性,以防止细胞粘附到内壁上。或者,抗细胞粘附的材料可用作第一细胞簇溶液发送通道51的材料。另外,具有高测温传导率的co2可渗透材料可用作第一细胞簇溶液发送通道51的材料,使得第一细胞簇溶液发送通道51的内部条件等同于容器200内的受控温度和co2浓度。根据防止污染的观点,第一细胞簇溶液发送通道51还可设置有逆流防止阀。
[0221]
第一细胞簇溶液发送通道51连接到第一分割机构60。第一分割机构60包括例如网格。当通过液压穿过网格时,溶液中包含的细胞簇被分割成与网格的每个孔隙的大小对应的多个细胞簇。例如,当网格具有均匀尺寸的孔隙时,因此分割的多个细胞簇的大小也几乎是均匀的。或者,第一分割机构60可包括喷嘴。例如,大体圆锥形的喷嘴的内部以阶梯形式微制造。当流过喷嘴时,溶液中包含的细胞簇被分割成多个细胞簇。扩增培养装置70连接到第一分割机构60。含有由第一分割机构60分割的细胞簇的溶液被送到扩增培养装置70。
[0222]
扩增培养装置70可在其中容纳孔板。扩增培养装置70还包括移液机。扩增培养装置70从第一分割机构60接收含有多个细胞簇的溶液,并通过移液机将溶液分配到孔中。在将细胞簇分配到孔中之后,扩增培养装置70例如在5%co2环境中在37℃下培养细胞簇大约八天。扩增培养装置70根据需要更换培养基。
[0223]
然后,扩增培养装置70向细胞簇添加作为胰蛋白酶的替代物的重组酶,诸如tryple select(r)(life technologies股份有限公司)。扩增培养装置70还将包含细胞簇的容器置于培养箱中,在培养箱中细胞簇与作为胰蛋白酶的替代物的重组酶在37℃下在5%co2环境中反应1分钟。然后,扩增培养装置70向含有细胞簇的溶液添加培养基,诸如维持培养基。扩增培养装置70还利用自动细胞刮刀等从容器中脱离细胞簇,并且经由扩增培养液发送通道71将含有细胞簇的溶液送入第一分割机构60。
[0224]
扩增培养装置70中的培养可在co2可渗透袋中而非孔板中进行。培养可为贴壁培养,或可为漂浮培养。或者,培养可为悬滴培养。在漂浮培养的情况下,可对培养进行搅拌。培养基可为琼脂形式。琼脂形式的培养基的示例包括结冷胶聚合物。当使用琼脂形式的培养基时,即使以漂浮培养的形式,不需要搅拌,因为细胞既不下沉也不粘附。
[0225]
扩增培养装置70可包括第二培养液补给装置,其对孔板或co2可渗透袋补给培养液。第二培养液补充装置可回收孔板或co2可渗透袋中的培养液,使用过滤器或透析膜过滤培养液,并循环纯化后的培养液。在这种情况下,可向待循环的培养液添加生长因子等。扩增培养装置70还可包括例如控制培养液温度的温度控制装置和控制培养液附近湿度的湿度控制装置。
[0226]
另外,在扩增培养装置70中,例如,如图4所示,可将细胞置于诸如透析膜的培养液可渗透袋301中,并且可将培养液可渗透袋301置于培养液不可渗透且co2可渗透的袋302中,而培养液可置于袋301和302中。可制备多个包含新鲜培养液的袋302,并且扩增培养装置70以预定的时间间隔用另一包含新鲜培养液的袋302替换其中置入包含细胞的袋301的袋302。扩增培养装置70中的培养方法不限于上述的方法,培养可通过后文提及的第二和第三实施例中描述的方法来进行。
[0227]
干细胞制造系统还可包括拍摄扩增培养装置70中的培养的扩增培养摄影装置。注意,当无色培养液用作扩增培养装置70中使用的培养液时,有可能抑制使用有色培养液时可发生的漫反射或自发荧光。因为诱导细胞和未诱导细胞在细胞形状和大小等方面不同,所以干细胞制造系统还可包括诱导状态监控装置,其通过拍摄扩增培养装置70中的细胞来计算诱导细胞的百分比。或者,诱导状态监控装置可通过抗体免疫染色方法或rna提取方法来鉴定诱导细胞的百分比。干细胞制造系统还可包括未诱导细胞除去装置,其通过磁性细胞分离方法、流式细胞术等来除去未诱导细胞。
[0228]
在扩增培养装置70中再次培养由第一分割机构60分割的细胞簇。重复第一分割机构60中对细胞簇的分割和扩增培养装置70中对细胞簇的培养,直到获得所需量的细胞。
[0229]
第二细胞簇溶液发送通道72连接到扩增培养装置70。扩增培养装置70使用泵等将含有从容器脱离的扩增培养后的细胞簇的溶液送入第二细胞簇溶液发送通道72。第二细胞簇溶液发送通道72可连接到分支通道,该分支通道具有仅允许具有小于预定大小的诱导细胞穿过并除去具有预定大小或更大的未诱导细胞的内径。
[0230]
第二细胞簇溶液发送通道72的内壁可通过涂覆聚hema而对细胞无粘附性,以防止细胞粘附到内壁上。或者,抗细胞粘附的材料可用作第二细胞簇溶液发送通道72的材料。另外,具有高测温传导率的co2可渗透材料可用作第二细胞簇溶液发送通道72的材料,使得第二细胞簇溶液发送通道72的内部条件等同于容器200内的受控温度和co2浓度。根据防止污染的观点,第二细胞簇溶液发送通道72还可设置有逆流防止阀。
[0231]
第二细胞簇溶液发送通道72连接到第二分割机构80。第二分割机构80包括例如网格。当通过液压穿过网格时,溶液中包含的细胞簇被分割成与网格的每个孔隙的大小对应的多个细胞簇。例如,当网格具有均匀尺寸的孔隙时,因此分割的多个细胞簇的大小也几乎是均匀的。或者,第二分割机构80可包括喷嘴。例如,大体圆锥形的喷嘴的内部以阶梯形式微制造。当流过喷嘴时,溶液中包含的细胞簇被分割成多个细胞簇。
[0232]
将多个细胞簇依次送入包装装置100的细胞簇输送机构90连接到第二分割机构80。预包装细胞通道91连接在细胞簇输送机构90与包装装置100之间。细胞簇输送机构90使用泵等将由第二分割机构80分割的细胞簇经由预包装细胞通道91依次送入包装装置100。
[0233]
预包装细胞通道91可用聚-hema涂覆以防止细胞粘附到其上。或者,抗细胞粘附的材料可用作预包装细胞通道91的材料。另外,具有高测温传导率的co2可渗透性材料可用作
预包装细胞通道91的材料,使得预包装细胞通道91的内部条件等同于容器200内的受控温度和co2浓度。根据防止污染的观点,预包装细胞通道91还可设置有逆流防止阀。
[0234]
冷冻保存液发送机构110连接到预包装细胞通道91。冷冻保存液发送机构110将细胞冷冻保存液送入预包装细胞通道91。因此,细胞簇悬浮在预包装细胞通道91中的细胞冷冻保存液中。
[0235]
包装装置100依次冷冻经由预包装细胞通道91发送的多个细胞簇。例如,每次接收到细胞簇时,包装装置100将细胞簇置于cryotube中,并立即冷冻细胞簇溶液,例如在-80℃或更低温度下。如果使用具有较小的单位体积表面积的cryotube,那么冷冻往往是耗时的。因此,优选地使用具有较大单位体积表面积的cryotube。通过使用具有较大单位体积表面积的cryotube有可能提高解冻后细胞的存活率。cryotube的形状的示例包括但不限于毛细管和球形。根据所需的解冻后细胞存活率,瞬间冷冻不是必需的。
[0236]
例如,在冷冻中使用玻璃化方法。在这种情况下,dap213(cosmo bio有限责任公司)或冷冻培养基(reprocell股份有限公司)可用作细胞冷冻保存液。可通过玻璃化方法以外的常规方法进行冷冻。在这种情况下,cryodefend-stem cell(r&d systems股份有限公司)、stem-cellbanker(r)(nippon zenyaku kogyo有限责任公司)等可用作细胞冷冻保存液。冷冻可使用液氮进行,或者可使用peltier设备进行。通过使用peltier设备,有可能调控温度改变并抑制温度变化。包装装置100将cryotube输出到容器200的外部。在临床使用冷冻细胞的情况下,cryotube优选地是完全密封的系统。然而,包装装置100可将干细胞包装在cryotube中而不冷冻干细胞。
[0237]
干细胞制造系统还可包括拍摄包装装置100中的包装步骤的包装步骤拍摄装置。
[0238]
干细胞制造系统还可包括对容器200的内部进行灭菌的灭菌装置。灭菌装置可为干热灭菌装置。注意,诸如分离装置10、转移前细胞溶液发送通道20、诱导物溶液发送机构21、诱导物转移装置30、细胞簇生产装置40和包装装置100的用电装置的线路优选地是具有耐热性的线路。或者,灭菌装置可通过向容器200内吹入臭氧气体、过氧化氢气体、福尔马林气体等灭菌气体来对容器200的内部进行灭菌。
[0239]
干细胞制造系统可通过有线或无线方式向外部服务器传输分离装置10、转移前细胞溶液发送通道20、诱导物溶液发送机构21、诱导物转移装置30、细胞簇生产装置40和包装装置100的操作记录以及由摄影装置拍摄的图像。外部服务器可使用神经网络来分析例如条件(例如,诱导物转移条件、培养条件和冷冻条件)与结果(例如,干细胞的不完全重编程、干细胞分化和增殖的失败以及染色体畸变)之间的联系,以提取导致结果的条件或预测结果。外部服务器还可基于标准操作程序(sop)调控干细胞制造系统中的分离装置10、诱导物溶液发送机构21、诱导物转移装置30、细胞簇生产装置40和包装装置100等,监控是否基于sop操作每个装置,并自动制作每个装置的操作记录。
[0240]
上述的干细胞制造系统有可能全自动地一次性实现诸如ips细胞的干细胞的诱导、建立、扩增培养和冷冻保存。
[0241]
(其他实施例)
[0242]
例如,诱导物转移装置30可通过rna转染来诱导细胞,而非通过电穿孔。或者,细胞可通过病毒(例如,逆转录病毒、慢病毒和仙台病毒)载体或质粒来诱导。转移前细胞溶液发送通道20、转移后细胞溶液发送通道31、细胞簇溶液发送通道51、扩展培养液发送通道71、
细胞簇溶液发送通道72和预包装细胞通道91可通过微流体技术设置在基底上。通过脂质转染将诱导物核糖核酸(rna)转移到诱导干细胞中以将干细胞分化成体细胞的装置可连接到干细胞制造系统。例如,后述的第六实施例中描述的方法可用作通过脂质转染将诱导物核糖核酸(rna)转移到诱导干细胞中以将干细胞分化成体细胞的方法。体细胞可为例如神经元细胞。
[0243]
(第二实施例)
[0244]
根据本发明第二实施例的用于干细胞的漂浮培养方法包括在凝胶培养基中漂浮培养干细胞。干细胞是例如诱导多能干(ips)细胞或胚胎干细胞(es细胞)。不搅拌凝胶培养基。凝胶培养基不含饲养细胞。干细胞在凝胶培养基中增殖,同时保持其未分化状态。
[0245]
例如,在漂浮培养之前,将干细胞解离成单细胞,并将解离成单细胞的干细胞置于凝胶培养基中。单细胞增殖,同时保持其克隆性,以在凝胶培养基中形成集落。
[0246]
凝胶培养基例如通过将脱乙酰结冷胶以0.5重量%至0.001重量%、0.1重量%至0.005重量%或0.05重量%至0.01重量%的终浓度添加至用于干细胞的培养基中来制备。
[0247]
凝胶培养基可含有选自由结冷胶、透明质酸、鼠李胶、定优胶、黄原胶、角叉菜胶、岩藻依聚糖、果胶、果胶酸、果胶酯酸、硫酸乙酰肝素、肝素、硫酸肝素、硫酸角质素、硫酸软骨素、硫酸皮肤素、硫酸鼠李聚糖及其盐组成的组的至少一种聚合物化合物。凝胶培养基可含有甲基纤维素。其中所含的甲基纤维素抑制细胞之间的聚集。
[0248]
或者,凝胶培养基可含有选自以下各项中的至少一种温敏性凝胶:聚(甘油单甲基丙烯酸酯)(pgma)、聚(2-羟丙基甲基丙烯酸酯)(phpma)、聚(n-异丙基丙烯酰胺)(pnipam)、胺基封端的、羧酸封端的、马来酰亚胺封端的、n-羟基丁二酰亚胺(nhs)酯封端的、三乙氧基硅烷封端的、聚(n-异丙基丙烯酰胺-共-丙烯酰胺)、聚(n-异丙基丙烯酰胺-共-丙烯酸)、聚(n-异丙基丙烯酰胺-共-丙烯酸丁酯)、聚(n-异丙基丙烯酰胺-共-甲基丙烯酸)、聚(n-异丙基丙烯酰胺-共-甲基丙烯酸-共-十八烷基丙烯酸酯)和n-异丙基丙烯酰胺。
[0249]
人es/ips培养基,例如primate es细胞培养基(reprocell股份有限公司)可用作用于干细胞的培养基。
[0250]
然而,用于干细胞的培养基不限于此,可使用各种干细胞培养基。例如,可使用primate es细胞培养基、reprostem、reproff、reproff2、reproxf(reprocell股份有限公司)、mtesr1、tesr2、tesre8、reprotesr(stemcell technologies股份有限公司)、pluristem (r)人es/ips培养基(merck kgaa)、用于人ips和es细胞的nutristem(r)xf/ff培养基、pluriton重编程培养基(stemgent股份有限公司)、pluristem(r)、stemfit ak02n、stemfit ak03(ajinomoto股份有限公司)、用于hesc/ips的无血清和饲养细胞培养基esc-sure(r)(applied stemcell股份有限公司)和l7(r)hpsc培养系统(lonza japan有限责任公司)。
[0251]
例如,每天将rock抑制剂以1000μmol/l或更高和0.1μmol/l或更低、100μmol/l或更高和1μmol/l或更低,或5μmol/l或更高和20μmol/l或更低的终浓度添加到凝胶培养基。添加至凝胶培养基的rock抑制剂促进干细胞形成集落。
[0252]
凝胶培养基可例如不含诸如碱性成纤维细胞生长因子(bfgf)的生长因子。或者,凝胶培养基可含有浓度为400μg/l或更低、40μg/l或更低或10μg/l或更低的低浓度的诸如bfgf的生长因子。与含有高浓度的诸如bfgf的生长因子的凝胶培养基相比,不含诸如bfgf
的生长因子的凝胶培养基或含有低浓度的诸如bfgf的生长因子的凝胶培养基容易促进干细胞的集落形成。
[0253]
凝胶培养基可不含tgf-β,或者可含有浓度为600ng/l或更低、300ng/l或更低或100ng/l或更低的低浓度的tgf-β。
[0254]
凝胶培养基中干细胞的浓度可为例如2
×
105个细胞/ml或更高、2.25
×
105个细胞/ml或更高,或2.5
×
105个细胞/ml或更高。如果凝胶培养基中干细胞的浓度低于2
×
105个细胞/ml,那么集落形成率趋于降低。
[0255]
根据本发明第二实施例的用于干细胞的漂浮培养方法有可能从单细胞形成干细胞集落。虽然这种方法是一种漂浮培养方法,但是由于干细胞的漂浮培养方法不需要搅拌培养基,因此干细胞不相互碰撞。因此,有可能保持集落的克隆性。因此,当干细胞是例如ips细胞时,可确保源自一个体细胞的ips细胞的克隆性。另外,由于干细胞不相互碰撞,因此干细胞集落可保持均匀的大小。此外,与贴壁培养方法相比,漂浮培养方法可在小空间内培养大量的集落。注意,可在漂浮培养中维持培养干细胞簇。
[0256]
自发现es/ips细胞以来,诸如bfgf或tgf-β的生长因子一直认为是es/ips细胞的培养所必需的。然而,诸如bfgf的生长因子在大约37℃的培养条件下迅速分解。因此,需要每天用新鲜的含有bfgf或tgf-β的培养液来更换含有bfgf或tgf-β的培养液,或者每天向其中添加bfgf或tgf-β等。用于培养的bfgf通常是重组蛋白。临床等级的重组蛋白需要依据非常严格的条例来生产。
[0257]
当bfgf的浓度例如为10ng/ml的低浓度时,一直认为需要将源自小鼠的成纤维细胞用作饲养细胞。然而,利用源自诸如小鼠的动物的饲养细胞培养的干细胞不可用于移植或再生医学。这一直是干细胞临床应用的瓶颈。
[0258]
虽然也开发了不使用饲养细胞的无饲养细胞的培养液,但是无饲养细胞培养液通常含有其量为使用饲养细胞的培养液中所含的量的25倍的bfgf并且含有浓度非常高的bfgf,诸如100ng/ml。然而,使用含有高浓度bfgf的无饲养细胞培养液难以培养没有核型异常的es/ips细胞。因此,许多ips细胞被破坏。在含有高浓度bfgf的无饲养细胞培养液中培养的es/ips细胞往往不太可能分化成特定的体细胞。因此,无饲养细胞培养液部分导致了从干细胞移植所必需的体细胞的生产效率的降低。
[0259]
相比之下,根据本发明第二实施例的用于干细胞的漂浮培养方法有可能在不使用饲养细胞的情况下培养和增殖干细胞,同时保持其未分化状态。即使不使用饲养细胞,根据本发明第二实施例的用于干细胞的漂浮培养方法也有可能在不使用诸如bfgf的生长因子或者使用低浓度的诸如bfgf的生长因子的情况下培养和增殖干细胞,同时保持其未分化状态。
[0260]
(第三实施例)
[0261]
如图5所示,根据本发明第三实施例的用于干细胞的漂浮培养容器包括:容纳干细胞和凝胶培养基的透析管;以及容纳透析管的容器,其中凝胶培养基置于透析管周围。
[0262]
透析管对例如rock抑制剂是可渗透的。透析管的截留分子量为0.1kda或更大、10kda或更大,或50kda或更大。透析管由例如纤维素酯、乙基纤维素、纤维素酯衍生物、再生纤维素、聚砜、聚丙烯腈、聚甲基丙烯酸甲酯、乙烯-乙烯醇共聚物、聚酯基聚合物合金、聚碳酸酯、聚酰胺、醋酸纤维素、二醋酸纤维素、三醋酸纤维素、铜铵人造丝、皂化纤维素、血仿透
析膜、磷脂酰胆碱膜或维生素e包膜制成。诸如离心管的圆锥管可用作容器。容器由例如聚丙烯制成。
[0263]
待置于透析管中的干细胞与第二实施例中的相同。同样,待置于透析管中的凝胶培养基与第二实施例中的相同。然而,待置于透析管中的凝胶培养基可不含rock抑制剂。待置于容器中透析管周围的凝胶培养基与第二实施例中的相同。待置于容器中透析管周围的凝胶培养基含有rock抑制剂。
[0264]
在透析管中干细胞的漂浮培养过程中,用新鲜凝胶培养基更换或补充容器中透析管周围的凝胶培养基。然而,更换透析管中的凝胶培养基可为不必要的。
[0265]
对于常规的漂浮培养系统,可难以在不吸取细胞的情况下更换培养基。然而,除非用新鲜培养基更换培养基,否则废物可积累。此外,除非用新鲜培养基更换或补充培养基,否则培养基可缺少培养基组分。
[0266]
相比之下,通过使用根据本发明第三实施例的用于干细胞的漂浮培养容器,即使用新鲜培养基更换透析管周围的培养基,也有可能避免吸取干细胞,因为干细胞在透析管中。此外,即使当用新鲜培养基更换透析管周围的培养基时,透析管中的培养基的量也难以改变。因此,透析管中的干细胞的密度不变。透析管中高浓度的废物移动到透析管的外部。培养基组分从透析管的外部培养基移动到透析管中,其中透析管中培养基组分的浓度降低。因此,有可能将干细胞周围的培养基保持新鲜。
[0267]
(第四实施例)
[0268]
根据本发明第四实施例的诱导干细胞的方法包括从在凝胶培养基中漂浮培养的体细胞诱导干细胞。体细胞是例如成纤维细胞。干细胞是例如ips细胞。不搅拌凝胶培养基。凝胶培养基不含饲养细胞。
[0269]
凝胶培养基例如通过将脱乙酰结冷胶以0.5重量%至0.001重量%、0.1重量%至0.005重量%或0.05重量%至0.01重量%的终浓度添加至用于干细胞的培养基来制备。
[0270]
凝胶培养基可含有选自由结冷胶、透明质酸、鼠李胶、定优胶、黄原胶、角叉菜胶、岩藻依聚糖、果胶、果胶酸、果胶酯酸、硫酸乙酰肝素、肝素、硫酸肝素、硫酸角质素、硫酸软骨素、硫酸皮肤素、硫酸鼠李聚糖及其盐组成的组的至少一种聚合物化合物。凝胶培养基可以含有甲基纤维素。其中所含的甲基纤维素抑制细胞之间的聚集。
[0271]
或者,凝胶培养基可含有选自以下各项中的至少一种温敏性凝胶:聚(甘油单甲基丙烯酸酯)(pgma)、聚(2-羟丙基甲基丙烯酸酯)(phpma)、聚(n-异丙基丙烯酰胺)(pnipam)、胺基封端的、羧酸封端的、马来酰亚胺封端的、n-羟基丁二酰亚胺(nhs)酯封端的、三乙氧基硅烷封端的、聚(n-异丙基丙烯酰胺-共-丙烯酰胺)、聚(n-异丙基丙烯酰胺-共-丙烯酸)、聚(n-异丙基丙烯酰胺-共-丙烯酸丁酯)、聚(n-异丙基丙烯酰胺-共-甲基丙烯酸)、聚(n-异丙基丙烯酰胺-共-甲基丙烯酸-共-十八烷基丙烯酸酯)和n-异丙基丙烯酰胺。
[0272]
人es/ips培养基,例如primate es细胞培养基(reprocell股份有限公司)可用作用于干细胞的培养基。
[0273]
然而,用于干细胞的培养基不限于此,可使用各种干细胞培养基。例如,可使用primate es细胞培养基、reprostem、reproff、reproff2、reproxf(reprocell股份有限公司)、mtesr1、tesr2、tesre8、reprotesr(stemcell technologies股份有限公司)、pluristem (r)人es/ips培养基(merck kgaa)、用于人ips和es细胞的nutristem(r)xf/ff
培养基、pluriton重编程培养基(stemgent股份有限公司)、pluristem(r)、stemfit ak02n、stemfit ak03(ajinomoto股份有限公司)、用于hesc/ips的无血清和饲养细胞培养基esc-sure(r)(applied stemcell股份有限公司)和l7(r)hpsc培养系统(lonza japan有限责任公司)。将凝胶培养基置于例如管中。
[0274]
凝胶培养基可例如不含诸如碱性成纤维细胞生长因子(bfgf)的生长因子。或者,凝胶培养基可含有浓度为400μg/l或更低、40μg/l或更低或10μg/l或更低的低浓度的诸如bfgf的生长因子。
[0275]
凝胶培养基可不含tgf-β,或者可含有浓度为600ng/l或更低、300ng/l或更低或100ng/l或更低的低浓度的tgf-β。
[0276]
(实例1)
[0277]
将500ml的primate es细胞培养基(reprocell股份有限公司)和0.2ml的浓度为10μg/ml的bfgf(gibco phg0266)混合以制备含有bfgf的人ips培养基。
[0278]
将脱乙酰结冷胶(nissan chemical industries有限责任公司)以0.02重量%的浓度添加至含有bfgf的人ips培养基以制备含有bfgf的人ips凝胶培养基。另外,将5ml的浓度为2.5重量%的胰蛋白酶、5ml的浓度为1mg/ml的胶原酶iv、0.5ml的浓度为0.1mol/l的cacl2、10ml的knockout serum replacement(r)(invitrogen 10828-028)和30ml的纯化水混合以制备通常称为ctk溶液的解离溶液。
[0279]
在饲养细胞上培养的过程中,将ctk溶液以300μl/孔添加至含有ips细胞的6孔培养皿(thermo fisher scientific 12-556-004),并将6孔培养皿在co2培养箱中温育3分钟。三分钟之后,将培养皿从培养箱中取出。确认仅饲养细胞从培养皿上脱离后,用抽吸器除去ctk溶液。在除去ctk溶液之后,通过以500μl/孔向6孔培养皿添加pbs(santa cruz biotech sc-362183)来洗涤ips细胞,随后从6孔培养皿中除去pbs。将解离溶液(accutase(r))以0.3ml/孔添加至6孔培养皿,然后将其置于co2培养箱中温育5分钟。然后,将含有bfgf的ips培养基以0.7ml/孔添加至6孔培养皿,使得ips细胞悬浮直到变成单细胞。
[0280]
在ips细胞悬浮之后,将4ml的含有bfgf的人ips培养基添加至15ml的离心管,并使用离心机以270g离心ips细胞悬浮液。在离心之后,除去上清液,向15ml的离心管添加1ml的含有bfgf的人ips培养基。使用血细胞计数器计算细胞的数量。在细胞计数后,将5
×
105个ips细胞接种到15ml的falcon管(corning 352096)或非粘附培养皿中,随后在不搅拌的情况下漂浮培养。
[0281]
在15ml的管中,使用2ml的含有bfgf的人ips凝胶培养基。在非粘附培养皿中,使用2ml的含有bfgf且不含结冷胶的人ips培养基。以10μmol/l向每个培养基添加rock抑制剂(selleck chemicals s1049)。然后,每天将500μl的含有bfgf的人ips凝胶培养基添加至15ml的管和非粘附培养皿,并且每天将500μl的含有bfgf的人ips培养基添加至非粘附培养皿。此外,每天将rock抑制剂以10μmol/l的终浓度添加至15ml的管和非粘附培养皿,并且持续漂浮培养7天。
[0282]
结果如图6所示。如图6(b)所示,在使用含有bfgf且不含结冷胶的人ips培养基在非粘附培养皿中培养ips细胞时,明显观察到了ips细胞集落之间的聚集。相比之下,如图6(a)所示,在使用含有bfgf的人ips凝胶培养基在15ml的管中培养ips细胞时,未观察到明显的聚集。图7(a)是当使用含有bfgf的人ips凝胶培养基在15ml的管中培养ips细胞时,第1天
拍摄的照片。图7(b)是当使用含有bfgf的人ips凝胶培养基在15ml的管中培养ips细胞时,第9天拍摄的照片。根据图7(a)和图7(b)的照片,确认不同系的ips细胞形成了它们各自的集落而不相互聚集。
[0283]
图8(a)是在凝胶培养基中漂浮培养了7天的ips细胞的集落在饲养细胞上紧随重新接种之前拍摄的照片。图8(b)是在形态学上确认集落三天后拍摄的照片。因此,如图9所示,确认95%以上的集落未分化。这些结果表明,可以在凝胶培养基中培养ips细胞,同时保持其未分化状态。
[0284]
(实例2)
[0285]
制备与实例1相同的含有bfgf的人ips培养基和含有bfgf的人ips凝胶培养基。在饲养细胞上培养的过程中,将ctk溶液以300μl/孔添加至含有ips细胞的6孔培养皿,并将6孔培养皿在co2培养箱中温育3分钟。三分钟之后,将培养皿从培养箱中取出。确认仅饲养细胞从培养皿上脱离后,用抽吸器除去ctk溶液。在除去ctk溶液之后,通过以500μl/孔向培养皿添加pbs来洗涤ips细胞,随后从培养皿中除去pbs。将accumax以0.3ml/孔添加至培养皿,然后将其置于co2培养箱中温育5分钟。然后,将含有bfgf的ips培养基以0.7ml/孔添加至培养皿,使得ips细胞悬浮直到变成单细胞。
[0286]
在ips细胞悬浮之后,将4ml的含有bfgf的人ips培养基添加至15ml的离心管中,并使用离心机以270g离心ips细胞悬浮液。在离心之后,除去上清液,向15ml的离心管中添加1ml的含有bfgf的人ips培养基。使用血细胞计数器计算细胞的数量。在细胞计数之后,将5
×
105个ips细胞接种到15ml的管中,随后在不搅拌的情况下进行漂浮培养。
[0287]
在15ml的管中,使用2ml的含有bfgf的人ips凝胶培养基。以10μmol/l向每个培养基中添加rock抑制剂。然后,每天将500μl的含有bfgf的人ips凝胶培养基添加至15ml的管。该凝胶培养基(500μl)含有0.5μl的rock抑制剂。作为对照,除了不添加rock抑制剂之外,在与上述相同的条件下将ips细胞漂浮培养7天。
[0288]
如图10(a)所示,当rock抑制剂不添加至含有bfgf的人ips培养基时,ips细胞不形成集落。相比之下,如图10(b)所示,当rock抑制剂添加至含有bfgf的人ips培养基时,ips细胞形成集落。这些结果表明,rock抑制剂对于从单细胞中漂浮培养ips细胞是有效的。
[0289]
(实例3)
[0290]
以与实例1中相同的方式制备含有bfgf的人ips凝胶培养基。另外,制备不含bfgf的人ips培养基,除不含bfgf之外与含有bfgf的人ips培养基相同。另外,制备不含bfgf的人ips凝胶培养基,除不含bfgf之外与含有bfgf的人ips凝胶培养基相同。此外,将脱酰结冷胶(nissan chemical industries有限责任公司)以0.02重量%的浓度添加至市售的无血清、无异源且无饲养细胞的用于重编程的培养基以制备用于对比的凝胶培养基。
[0291]
在此,含有bfgf的人ips凝胶培养基仅含有浓度为用于对比的凝胶培养基中bfgf浓度的大约1/25的bfgf。
[0292]
在饲养细胞上培养的过程中,将ctk溶液以300μl/孔添加至含有ips细胞的6孔培养皿中,并将6孔培养皿在co2培养箱中温育3分钟。三分钟之后,将培养皿从培养箱中取出。在确认仅饲养细胞从培养皿上脱离之后,用抽吸器除去ctk溶液。在除去ctk溶液之后,将ips细胞用pbs洗涤一次。向其添加1ml的不含bfgf的人ips培养基,并且使用刮刀刮取ips细胞,并将其悬浮在15ml的离心管中约10次以不变成单细胞。然后,向其添加2ml的不含bfgf
的人ips培养基,并且将混合物分成3等份,每份1ml,将这3等份使用离心机以270g离心。
[0293]
在离心之后,从15ml的离心管中除去上清液,向15ml的离心管添加2ml的用于对比的凝胶培养基、含有bfgf的人ips凝胶培养基或不含有bfgf的人ips凝胶培养基。从第二天起,每天向离心管添加500μl的与最初凝胶培养基相同的凝胶培养基,并将ips细胞漂浮培养七天。
[0294]
图11(a)示出了在从市售无饲养细胞培养基制备的用于对比的凝胶培养基中漂浮培养7天的ips细胞的集落的典型示例。图11(b)示出了在含有bfgf的人ips凝胶培养基中漂浮培养七天的ips细胞的集落的典型示例。图11(c)示出了在不含bfgf的人ips凝胶培养基中漂浮培养7天的ips细胞的集落的典型示例。
[0295]
甚至在不含bfgf的人ips凝胶培养基和仅含有浓度为用于对比的凝胶培养基中bfgf浓度的大约1/25的bfgf的含有bfgf的人ips凝胶培养基中可培养ips细胞。
[0296]
为了确认漂浮培养7天的ips细胞的集落是否未分化,在饲养细胞上再次接种ips细胞,并对其集落进行形态学观察。图12的上部照片各自示出了凝胶培养基中的集落。图12的中间照片各自示出了在饲养细胞上重新接种漂浮培养7天的ips细胞之后两天的集落。在每种情况下,确认未分化的集落占90%或更多。这些结果表明,即使在使用不含bfgf的凝胶培养基或相比于用于对比的凝胶培养基的bfgf浓度低25倍或以上的凝胶培养基时,ips细胞可进行漂浮培养同时保持其分化状态。
[0297]
图12下部的照片示出了在饲养细胞上重新接种漂浮培养七天的ips细胞之后七天的集落。这些结果表明,即使在凝胶培养基中漂浮培养然后在饲养细胞上长时间(7天)培养,ips细胞未分化。
[0298]
(实例4)
[0299]
制备与实例3中相同的不含bfgf的人ips培养基、含有bfgf的人ips凝胶培养基和不含bfgf的人ips凝胶培养基。另外,将脱酰结冷胶(nissan chemical industries有限责任公司)以0.02重量%的浓度添加至市售无血清且无饲养细胞的培养基中以制备用于对比的凝胶培养基。以10μmol/l的浓度向所有凝胶培养基添加rock抑制剂。
[0300]
在饲养细胞上培养的过程中,将ctk溶液以300μl/孔添加至含有ips细胞的6孔培养皿,并将6孔培养皿在co2培养箱中温育3分钟。三分钟之后,将培养皿从培养箱中取出。在确认仅饲养细胞从培养皿上脱离后,用抽吸器除去ctk溶液。在除去ctk溶液之后,通过以500μl/孔向6孔培养皿添加pbs来洗涤ips细胞,随后从6孔培养皿中除去pbs。将accumax以0.3ml/孔添加至6孔培养皿,然后将其置于co2培养箱中温育5分钟。然后,将含有bfgf的ips培养基以0.7ml/孔添加至6孔培养皿,使得ips细胞悬浮直到变成单细胞。
[0301]
在ips细胞悬浮之后,将4ml的不含bfgf的人ips培养基添加至15ml的离心管,并使用离心机以270g离心ips细胞悬浮液。在离心之后,除去上清液,向离心管中添加1ml的不含bfgf的人ips培养基。使用血细胞计数器计算细胞的数量。
[0302]
然后,每个离心管置入5
×
105个ips细胞,向离心管添加2ml的含有bfgf的人ips凝胶培养基、不含bfgf的人ips凝胶培养基或用于对比的凝胶培养基。从第二天起,每天向离心管添加500μl的与最初凝胶培养基相同的凝胶培养基,并将ips细胞漂浮培养七天。
[0303]
因此,如图13(c)所示,源自单细胞的ips细胞不能在用于对比的凝胶培养基中培养。相比之下,如图13(a)和图13(b)所示,源自单细胞的ips细胞可在含有bfgf的人ips凝胶
培养基和不含bfgf的人ips凝胶培养基中培养。含有bfgf的人ips凝胶培养基具有4μg/ml的bfgf浓度,用于对比的凝胶培养基具有100μg/ml的bfgf浓度。
[0304]
如图14所示,作为确定集落数的结果,在不含bfgf的人ips凝胶培养基中漂浮培养的ips细胞形成的集落数是在含有bfgf的人ips凝胶培养基中漂浮培养的ips细胞的集落数的两倍或更多。这些结果表明,对于凝胶培养基,优选的是低bfgf浓度或不存在bfgf。
[0305]
此外,将ips细胞解离成单细胞,并使用其中添加了rock抑制剂的培养基培养7天,rock抑制剂以10μmol/l添加至含有bfgf的人ips培养基或补充有0.02重量%的脱酰结冷胶的不含bfgf的人ips培养基。在该操作中,5
×
105个细胞悬浮在1.5ml的每种凝胶培养基中,并且每天向1.5ml的每种凝胶培养基以10μmol/l添加rock抑制剂。
[0306]
向培养7天的ips细胞添加10倍量的pbs。在使用离心机以270g离心之后,弃去上清液,向培养容器添加0.3ml的accumax,然后将其置于co2培养箱中温育5分钟。然后,向其添加0.7ml的含有bfgf的人ips培养基,使得ips细胞悬浮直到成为单细胞。在悬浮之后,向其添加1.5ml的人ips培养基(用于在含有bfgf的培养基中培养的细胞的含有bfgf的培养基,或用于在不含bfgf的培养基中培养的细胞的不含bfgf的培养基),并且将ips细胞以与前7天相同的方式使用离心机再培养7天。在培养之后,将等分试样重新接种在饲养细胞上。再过三天之后,用抗nanog和oct3/4的抗体将细胞染色并观察。结果如图15所示。在凝胶培养基中培养总共14天的ips细胞对于未分化标记nanog和oct3/4呈阳性。这些结果表明,即使在使用不含bfgf的凝胶培养基时,ips细胞可在凝胶培养基中长期培养,同时保持其未分化状态。
[0307]
(实例5)
[0308]
制备与实例3中相同的不含bfgf的人ips培养基和不含bfgf的人ips凝胶培养基。将rock抑制剂以10μmol/l的浓度添加至两种凝胶培养基。
[0309]
在饲养细胞上培养的过程中,将ctk溶液以300μl/孔添加至含有ips细胞的6孔培养皿,并将6孔培养皿在co2培养箱中温育3分钟。三分钟之后,将培养皿从培养箱中取出。在确认仅饲养细胞从培养皿上脱离之后,用抽吸器除去ctk溶液。在除去ctk溶液之后,通过以500μl/孔向6孔培养皿添加pbs来洗涤ips细胞,随后除去pbs。将accutase以0.3ml/孔添加至6孔培养皿,然后将其置于co2培养箱中温育5分钟。然后,将不含bfgf的人ips培养基以0.7ml/孔添加至6孔培养皿,使得ips细胞悬浮直到变成单细胞。
[0310]
在ips细胞悬浮之后,将4ml的不含bfgf的人ips培养基添加至离心管,并使用离心机以270g离心ips细胞悬浮液。在离心之后,除去上清液,向离心管添加1ml的不含bfgf的人ips培养基。使用血细胞计数器计算细胞的数量。
[0311]
然后,每个离心管置入1
×
105、2.5
×
105或5
×
105个ips细胞,并向其添加2ml的不含bfgf的人ips凝胶培养基。从第二天起,每天向离心管添加500μl的凝胶培养基,并将ips细胞漂浮培养七天。
[0312]
图16示出了各接种细胞数情况下的集落的照片。图17示出了确定形成集落的ips细胞的数量与接种ips细胞的数量的比率的结果。以5
×
105个细胞接种的ips细胞形成了数量为以1
×
105或2.5
×
105个细胞接种的ips细胞的集落数的10倍或更多倍的集落。这些结果表明,以低浓度接种的ips细胞不形成集落。
[0313]
每个离心管置入1
×
105个ips细胞,并向离心管添加200μl、400μl、1000μl或2000μ
l的不含bfgf的人ips凝胶培养基。从第二天起,每天向离心管添加100μl、200μl、5000μl或1000μl的凝胶培养基,并将ips细胞漂浮培养7天。
[0314]
图18示出了确定形成集落的ips细胞的数量与接种的ips细胞的数量的比率的结果。这些结果表明,随着凝胶培养基量的增加,换句话说,随着ips细胞的接种浓度的降低,ips细胞更不易形成集落。
[0315]
(实例6)
[0316]
制备与实例3中相同的不含bfgf的人ips培养基和不含bfgf的人ips凝胶培养基。
[0317]
在饲养细胞上培养的过程中,将ctk溶液以300μl/孔添加至含有ips细胞的6孔培养皿,并将6孔培养皿在co2培养箱中温育3分钟。三分钟之后,将培养皿从培养箱中取出。在确认仅饲养细胞从培养皿上脱离之后,用抽吸器除去ctk溶液。在除去ctk溶液之后,通过向6孔培养皿以500μl/孔添加pbs来洗涤细胞,随后除去pbs。将accumax以0.3ml/孔添加至6孔培养皿,然后将其置于co2培养箱中温育5分钟。然后,将不含bfgf的人ips培养基以0.7ml/孔添加至6孔培养皿,使得ips细胞悬浮直到变成单细胞。
[0318]
在ips细胞悬浮之后,将4ml的不含bfgf的人ips培养基添加至15ml的离心管,并使用离心机以270g离心ips细胞悬浮液。在离心之后,除去上清液,向15ml的离心管添加1ml的不含bfgf的人ips培养基。使用血细胞计数器计算细胞的数量。
[0319]
然后,将2ml的含有5
×
105个ips细胞的不含bfgf的人ips凝胶培养基置于配备有截留分子量为100kda的透析管的透析模块(spectrum laboratories g235035)中。透析管中未放置rock抑制剂。如图5所示,进一步将透析模块置于50ml的离心管中,并将20ml的不含bfgf的人ips凝胶培养基置于离心管中透析管周围。进一步将rock抑制剂以10μmol/l的终浓度添加至透析管周围的不含bfgf的人ips凝胶培养基。作为对照,未向一些离心管中添加rock抑制剂。然后,每两天用新鲜凝胶培养基更换10ml的透析管周围的不含bfgf的人ips凝胶培养基,并持续漂浮培养7天。待置入用于更换的不含bfgf的新鲜人ips凝胶培养基含有浓度为10μmol/l的rock抑制剂。
[0320]
如图20所示,与图19(a)所示的在将不含bfgf和rock抑制剂的人ips凝胶培养基置于透析管周围的情况下的培养相比,通过如图19(b)所示的在将补充有rock抑制剂的不含bfgf的人ips凝胶培养基置于透析管周围的情况下的培养,ips细胞显著形成了集落。
[0321]
这些结果表明诸如rock抑制剂的低分子穿过透析管的膜。这些结果还表明,可在维持透析管中培养基成分的浓度的同时培养ips细胞。
[0322]
(实例7)
[0323]
制备与实例3中相同的不含bfgf的人ips培养基和不含bfgf的人ips凝胶培养基。
[0324]
在饲养细胞上培养的过程中,将ctk溶液以300μl/孔添加至含有ips细胞的6孔培养皿,并将6孔培养皿在co2培养箱中温育3分钟。三分钟之后,将培养皿从培养箱中取出。在确认仅饲养细胞从培养皿上脱离之后,用抽吸器除去ctk溶液。在除去ctk溶液之后,通过以500μl/孔向6孔培养皿添加pbs来洗涤细胞,随后从6孔培养皿中除去pbs。将accumax以0.3ml/孔添加至6孔培养皿,然后将其置于co2培养箱中温育5分钟。然后,将不含bfgf的人ips培养基以0.7ml/孔添加至6孔培养皿,使得ips细胞悬浮直到变成单细胞。
[0325]
在ips细胞悬浮之后,将4ml的不含bfgf的人ips培养基添加至15ml的离心管,并使用离心机以270g离心ips细胞悬浮液。在离心之后,除去上清液,向离心管添加1ml的不含
bfgf的人ips培养基。使用血细胞计数器计算细胞的数量。
[0326]
然后,将2ml的含有5
×
105个ips细胞的不含bfgf的人ips凝胶培养基置于透析模块的透析管中。进一步将透析模块置于50ml的离心管(corning352070)中,并将20ml的不含bfgf的人ips凝胶培养基置于离心管中透析管周围。进一步将rock抑制剂以10μmol/l添加至透析管周围的不含bfgf的人ips凝胶培养基。然后,每两天用新鲜凝胶培养基更换10ml的透析管周围的不含bfgf的人ips凝胶培养基,并持续漂浮培养7天。待置入用于更换的不含bfgf的新鲜人ips凝胶培养基含有浓度为10μmol/l的rock抑制剂。作为对照,在一些离心管中持续漂浮培养七天,而无需更换透析管周围的不含bfgf的人ips凝胶培养基。
[0327]
如图22所示,与图21(b)所示的不更换透析管周围的不含bfgf的人ips凝胶培养基的情况相比,在图21(a)所示的用新鲜凝胶培养基更换透析管周围的不含bfgf的人ips凝胶培养基的情况下,发现ips细胞所形成的单个集落较大。这些结果表明,更换透析管周围的不含bfgf的人ips凝胶培养基促进了ips细胞增殖的能力。
[0328]
将漂浮培养7天的ips细胞的集落进一步重新接种在饲养细胞上,并根据集落的形态确认ips细胞的未分化状态得到维持。如图23和图24所示,即使更换或不更换透析管周围的不含bfgf的人ips凝胶培养基,80%或更多的集落保持其未分化状态。
[0329]
(实例8)
[0330]
制备与实例3中相同的不含bfgf的人ips培养基和不含bfgf的人ips凝胶培养基。
[0331]
在饲养细胞上培养的过程中,将ctk溶液以300μl/孔添加至含有ips细胞的6孔培养皿,并将培养皿在co2培养箱中温育3分钟。三分钟之后,将培养皿从培养箱中取出。在确认仅饲养细胞从培养皿上脱离之后,用抽吸器除去ctk溶液。在除去ctk溶液之后,通过以500μl/孔向6孔培养皿添加pbs来洗涤细胞,然后除去pbs。将accumax以0.3ml/孔添加至6孔培养皿,然后将其置于co2培养箱中温育5分钟。然后,将不含bfgf的人ips培养基以0.7ml/孔添加至6孔培养皿,使得ips细胞悬浮直到变成单细胞。
[0332]
在ips细胞悬浮之后,将4ml的不含bfgf的人ips培养基添加至15ml的离心管中,并使用离心机以270g离心ips细胞悬浮液。在离心之后,除去上清液,向离心管添加1ml的不含bfgf的人ips培养基。使用血细胞计数器计算细胞的数量。
[0333]
然后,将2ml的含有5
×
105个ips细胞的不含bfgf的人ips凝胶培养基置于透析模块的透析管中。进一步将透析模块置于50ml的离心管中,并将20ml的不含bfgf的人ips凝胶培养基置于离心管中透析管周围。进一步将rock抑制剂以10μmol/l添加至透析管周围的不含bfgf的人ips凝胶培养基。然后,每两天用新鲜凝胶培养基更换10ml的透析管周围的不含bfgf的人ips凝胶培养基,并持续漂浮培养7天。待置入用于更换的不含bfgf的新鲜人ips凝胶培养基含有浓度为10μmol/l的rock抑制剂。
[0334]
作为第一对照,将2ml的含有5
×
105个ips细胞的不含bfgf的人ips凝胶培养基置于透析模块的透析管中。进一步将透析管置于50ml的离心管中,并将20ml的不含bfgf和结冷胶的人ips培养基置于离心管中透析管周围。进一步将rock抑制剂以10μmol/l添加至透析管周围的不含bfgf和结冷胶的人ips培养基中。然后,每两天用新鲜培养基更换透析管周围的10ml的不含bfgf和结冷胶的人ips培养基,并持续漂浮培养7天。
[0335]
作为第二对照,将2ml的含有5
×
105个ips细胞的不含bfgf的人ips凝胶培养基置于50ml的离心管中而不使用透析管。然后,每天一次将500μl的不含bfgf的人ips凝胶培养
基添加至50ml的离心管,并持续漂浮培养七天。
[0336]
因此,如图25和图26所示,与不使用透析管相比,使用透析管增加了ips细胞的集落的数量。此外,与在透析管周围使用不含bfgf和结冷胶的人ips培养基相比,在透析管周围使用不含bfgf的人ips凝胶培养基增加了ips细胞的集落的数量。
[0337]
(实例9:在聚合物培养基中诱导ips细胞)
[0338]
将500ml的primate es细胞培养基(reprocell股份有限公司)和0.2ml的浓度为10μg/ml的bfgf(gibco phg0266)混合以制备含有bfgf的人ips培养基。另外,在不与bfgf(gibco phg0266)混合的情况下,从500ml的primate es细胞培养基(reprocell股份有限公司)制备不含bfgf的人ips培养基。另外,制备市售的无血清和无饲养细胞的培养基。
[0339]
将脱乙酰结冷胶(nissan chemical industries有限责任公司)以0.02重量%的浓度添加至不含bfgf的人ips培养基、含有bfgf的人ips培养基和市售的无血清培养基和无饲养细胞的培养基,以制备不含bfgf的人ips凝胶培养基、含有bfgf的人ips凝胶培养基和用于对比的凝胶培养基。
[0340]
使用逆转录病毒将oct3/4、sox2、klf4和c-myc转移到人成纤维细胞。在漂浮培养7天之后,1
×
105个细胞悬浮于不含bfgf的人ips凝胶培养基中,并将其在不含bfgf的人ips凝胶培养基、含有bfgf的人ips凝胶培养基或用于对比的凝胶培养基中进行培养。因此,生产出ips细胞。图示示于图27中。因此,在不含bfgf的人ips凝胶培养基中生产的ips细胞被重新接种在饲养细胞上。两天之后,在形态学上确认其集落,因此如图28(a)所示,该集落是未分化的ips细胞集落。如图28(b)和图28(c)所示,由于进一步用抗oct3/4和nanog的抗体将ips细胞染色,ips细胞对其呈阳性。这些结果表明可在聚合物培养基中诱导ips细胞。
[0341]
(实例10:在聚合物培养基中诱导的ips细胞的克隆性)
[0342]
在培养过程中,将300μl的ctk溶液添加至含有在聚合物培养基中诱导的ips细胞的6cm的培养皿,并将培养皿在co2培养箱中温育3分钟。三分钟之后,将培养皿从培养箱中取出。在确认仅饲养细胞从培养皿上脱离之后,用抽吸器除去ctk溶液。在除去ctk溶液之后,通过向培养皿添加500μl的pbs来洗涤ips细胞,随后除去pbs。向培养皿添加0.3ml的解离液(accumax),然后将其置于co2培养箱中温育5分钟。然后,将0.7ml的不含bfgf的ips培养基添加至培养皿,使得ips细胞悬浮直到变成单细胞。
[0343]
在ips细胞悬浮之后,将4ml的不含bfgf的ips培养基添加至离心管,并使用离心机以270g离心ips细胞悬浮液。在离心之后,除去上清液,向其添加1ml的不含bfgf的ips培养基。使用血细胞计数器计算细胞的数量。在细胞计数之后,使用cell explorer活细胞标记试剂盒red和细胞探测器活细胞标记试剂盒green(aat bioquest股份有限公司)将2.5
×
105个ips细胞染色。在染色之后,将染色后的细胞混合,并将5
×
105个ips细胞接种到非粘附培养皿或15ml的管中,随后在不搅拌的情况下漂浮培养。在15ml的管中,使用2ml的不含bfgf的人ips凝胶培养基。在非粘附培养皿中,使用2ml的不含bfgf和结冷胶的人ips培养基。将rock抑制剂(selleck chemicals s1049)以10μmol/l的浓度添加至每种培养基。然后,每天将500μl的不含bfgf的人ips培养基添加至15ml的管和非粘附培养皿。待置入用于更换的不含bfgf的新鲜人ips凝胶培养基含有浓度为10μmol/l的rock抑制剂。每天将rock抑制剂以10μmol/l的终浓度添加至15ml的管和非粘附培养皿,并持续漂浮培养七天。
[0344]
因此,如图29(a)所示,当在非粘附培养皿中使用不含结冷胶的培养基培养ips细
胞时,显著观察到了不同染色的ips细胞集落之间的聚集。作为定量的结果,40%或以上的细胞被聚集。相比之下,如图29(b)所示,当在15ml的管中使用不含bfgf的人ips凝胶培养基培养ips细胞时,未观察到此类聚集。
[0345]
(实例11)
[0346]
在ips细胞悬浮之后,将4ml的含有bfgf的人ips培养基添加至15ml的离心管,并使用离心机以270g离心ips细胞悬浮液。在离心之后,除去上清液,向15ml的离心管添加1ml的含有bfgf的人ips培养基。使用血细胞计数器计算细胞的数量。在细胞计数之后,将5
×
105个ips细胞接种到15ml的falcon管(r)(corning 352096)或非粘附培养皿中,随后在不搅拌的情况下进行漂浮培养。
[0347]
所使用的培养基是2ml的含有bfgf的人ips凝胶培养基或含有bfgf且不含结冷胶的人ips培养基,并将ips细胞在管或非粘附培养皿中培养5天至7天。将rock抑制剂(selleck chemicals s1049)以10μmol/l添加至每种培养基。然后,每天将500μl的含有bfgf和结冷胶的人ips培养基或含有bfgf且不含结冷胶的人ips培养基添加至15ml的管和非粘附培养皿。每天将rock抑制剂以10μmol/l的终浓度添加至15ml的管和非粘附培养皿,并持续漂浮培养5天至7天。
[0348]
图30(a)是示出在管中含有bfgf且不含结冷胶的人ips培养基中培养的ips细胞的照片。在这种情况下,ips细胞沉淀,因此无法培养。图30(b)是示出在管中含有bfgf和结冷胶的人ips培养基中培养的ips细胞的照片。在这种情况下,ips细胞既不沉淀也不聚集。图30(c)是示出在培养皿中含有bfgf且不含结冷胶的人ips培养基中培养的ips细胞的照片。在这种情况下,ips细胞聚集,因此不能进行培养。图30(d)是示出在培养皿中含有bfgf和结冷胶的人ips培养基中培养的ips细胞的照片。在这种情况下,ips细胞聚集,因此不能进行培养。
[0349]
(实例12)
[0350]
制备与实例3中相同的不含bfgf的人ips培养基和不含bfgf的人ips凝胶培养基。此外,制备市售的无血清和无饲养细胞的培养基。
[0351]
制备设置有形成网格图案的上部开口直径为0.8mm且下部开口直径为0.5mm的多个通孔的格栅板(spheroid generator mps 500和mpc 500,kuraray有限责任公司)。
[0352]
在饲养细胞上培养的过程中,将ctk溶液以300μl/孔添加至含有ips细胞的6孔培养皿,并将6孔培养皿在co2培养箱中温育3分钟。三分钟之后,将培养皿从培养箱中取出。在确认仅饲养细胞从培养皿上脱离之后,用抽吸器除去ctk溶液。在除去ctk溶液之后,通过以500μl/孔向培养皿添加pbs来洗涤细胞,随后除去pbs。将accumax以0.3ml/孔添加至培养皿,然后将其置于co2培养箱中温育5分钟。然后,将不含bfgf的人ips培养基以0.7ml/孔添加至培养皿,使得ips细胞悬浮直到变成单细胞。
[0353]
然后,将4ml的不含bfgf的人ips培养基添加至15ml的离心管,并使用离心机以270g离心ips细胞悬浮液。在离心之后,除去上清液,向离心管中添加1ml的不含bfgf的人ips培养基。使用血细胞计数器计算细胞的数量。
[0354]
然后,将2.5
×
105个ips细胞接种到每个格栅板上,并使用格栅板的每个通孔悬滴培养两天以形成如图31(a)所示的具有均匀大小的集落。接下来,将大小均匀的集落置于2ml的不含bfgf的人ips凝胶培养基中,并将包含集落的不含bfgf的人ips凝胶培养基置于
透析模块的透析管中。进一步将透析模块置于50ml的离心管中,并在离心管中透析管周围置入20ml的不含结冷胶的市售无血清且无饲养细胞的培养基。然后,每两天用新鲜培养基更换10ml的透析管周围的不含结冷胶的市售无血清且无饲养细胞的培养基,并持续漂浮培养7天。待置入用于更换的新鲜培养基含有浓度为10μmol/l的rock抑制剂。
[0355]
在漂浮培养7天后,如图31(b)和图32所示,观察到ips细胞集落的大小增加。这些结果表明ips细胞的集落增殖。
[0356]
进一步将漂浮培养的ips细胞集落重新接种在饲养细胞上。三天之后,从集落的形态确认ips细胞的未分化状态得到维持。因此,如图33和图34所示,所有的集落均未分化。这些结果表明,可在格栅板中使ips细胞集落的大小变得均匀,然后,可在聚合物培养基中培养ips细胞,同时保持其未分化状态。
[0357]
(第五实施例)
[0358]
根据本发明实施例的生产诱导多能干(ips)细胞的方法包括:制备体细胞;并通过脂质转染法将编码重编程因子的rna转移到体细胞中。
[0359]
体细胞是例如血细胞。血细胞从血液中分离。血液例如是外周血或脐带血,但血液不限于此。血液可从成年人身上采集,也可从未成年人身上采集。对于采血,使用诸如乙二胺四乙酸(edta)、肝素或柠檬酸葡萄糖配方a溶液(acd-a溶液)的抗凝剂。
[0360]
血细胞例如是有核细胞,诸如单核细胞、嗜中性粒细胞、嗜碱性粒细胞或淋巴细胞,并且不包括红细胞、粒细胞和血小板。血细胞可为例如血管内皮祖细胞、造血干/祖细胞、t细胞或b细胞。t细胞是例如αβt细胞。
[0361]
使用血细胞分离用培养基和离心分离装置等从血液中分离单核细胞。在将ficoll(ge healthcare japan股份有限公司)用作血细胞分离用培养基的情况下,分离单核细胞的方法如下。
[0362]
单核细胞分离准确性在低温下倾向于变差。因此,将离心机设定为4℃至42℃,优选18℃。从成人或未成年人采集10μl至50ml的血液,并将含有edta的螯合剂添加至血液以不使血液凝结,随后温和地混合。将人体淋巴细胞分离用培养基(ficoll-paque premium,ge healthcare japan股份有限公司)以5ml/管分配到两个15ml的管中。将5ml的pbs添加至5ml的血液进行稀释,并将5ml的稀释后的血液层积在各管中的人体淋巴细胞分离用培养基上。此时,将稀释后的血液缓慢地添加至培养基上,使得血液沿着管壁向下流动,从而不干扰分界面。
[0363]
将每管中的溶液在4℃至42℃,优选18℃下以10
×
g至1000
×
g,优选400
×
g离心5分钟至2小时,优选30分钟。在离心之后,管中出现白色混浊的中间层。该白色混浊的中间层中含有单核细胞。用pipetman逐渐回收管中的白色混浊的中间层,并将其转移到新15ml的管中。在这一操作中,有必要避免吸出下层。可从一个管中回收约1ml的量的白色混浊的中间层。将来自两个管的中间层一起转移到一个管。
[0364]
将1ml至48ml,优选12ml的pbs添加至回收的单核细胞,并进一步将溶液在4℃至42℃,优选18℃下以10
×
g至1000
×
g,优选200
×
g离心1分钟至60分钟,优选十分钟。然后,使用抽吸器吸去溶液的上清液,并通过添加1ml至12ml,优选3ml的具有已知组成的无血清造血细胞培养基(x-vivo(r)10,lonza japan有限责任公司)使单核细胞悬浮,以获得单核细胞悬浮液。将单核细胞悬浮液的10μl等分试样用trypan blue染色并使用血细胞计数器计
数。
[0365]
在vacutainer(r)(becton,dickinson and company)用作血液采集管的情况下,分离单核细胞的方法如下。
[0366]
单核细胞分离准确性在低温下倾向于变差。因此,离心机设定为4℃至42℃,优选18℃。使用血液采集管(vacutainer(r),becton,dickinson and company)从成人或未成年人采集8ml的血液,并通过倒置与抗凝血剂混合。然后,调节平衡,并使用摆动转子将溶液在4℃至42℃,优选18℃下,以100
×
g至3000
×
g,优选1500
×
g至1800
×
g离心1分钟至60分钟,优选20分钟。在离心之后,除去上层,即血浆层,通过吸移使单核细胞层和粘附于凝胶的血细胞悬浮,以获得悬浮液。将获得的悬浮液转移到另一个15ml的管。
[0367]
将1ml至14ml,优选12ml的pbs添加至15ml的管中的悬浮液,并将悬浮液在4℃至42
°
下,优选18℃下,以100
×
g至3000
×
g,优选200
×
g离心1分钟至60分钟,优选5分钟。在离心之后,使用抽吸器除去上清液。用无菌水将造血剂(pharmlyse(r),x10 concentrate,becton,dickinson and company)稀释至x1浓度。通过轻敲使15ml的管中的团粒解离,并向其添加1ml至14ml,优选1ml的造血剂。然后,将溶液在室温下在黑暗中静置1分钟至60分钟,优选1分钟。
[0368]
接下来,将1ml至14ml,优选12ml的pbs添加至15ml的管,并将溶液在4℃至42℃下,优选室温下,以100
×
g至3000
×
g,优选200
×
g,离心1分钟至60分钟,优选5分钟。在离心之后,用抽吸器除去上清液,并通过添加1ml至15ml,优选3ml的具有已知组成的无血清造血细胞培养基(x-vivo(r)10,lonza japan有限责任公司)使单核细胞悬浮,以得到单核细胞悬浮液。将单核细胞悬浮液的10μl等分试样用trypan blue染色并使用血细胞计数器计数。
[0369]
从血液中分离单核细胞的方法不限于上述方法,例如,可使用透析膜从血液中分离单核细胞。此外,可使用用于全血mnc富集的purecellselect系统(r)(pall股份有限公司)、用于除去血细胞的净化剂(cellsorba e(r),asahi kasei股份有限公司)和制作用于血小板浓缩物的白细胞除去过滤器(sepacell pl(r),plx-5b-scd,asahi kasei股份有限公司)等。
[0370]
购自cellular technology limited的ctl-up1、购自sanguine biosciences股份有限公司的pbmc-001等可用作单核细胞。
[0371]
或者,可解冻使用诸如cellbanker 1、gmp等级stem-cellbanker或不含dmso gmp等级stem-cellbanker(nippon zenyaku kogyo株式会社)的细胞冷冻保存液冷冻保存的血细胞并将其用作血细胞。
[0372]
为了解冻单核细胞,首先将1ml至15ml,优选8ml的具有已知组成的无血清造血细胞培养基(x-vivo(r)10,lonza japan有限责任公司)预先置于15ml的管中,并将含有冷冻单核细胞的管置于4℃至42℃,优选37℃的温浴中以开始单核细胞的解冻。然后,将仍有少量冰块的含有单核细胞的管从温浴中取出,并将单核细胞转移到含有具有已知组成的无血清造血细胞培养基的管。将单核细胞悬浮液的10μl等分试样用trypan blue染色并使用血细胞计数器计数。
[0373]
血细胞可以根据其细胞表面标记进行分离。造血干/祖细胞对于cd34呈阳性。t细胞对于cd3、cd4和cd8中的任何一种呈阳性。b细胞对于cd10、cd19和cd20中的任何一种呈阳性。使用例如自动磁性细胞分离装置将造血干细胞/祖细胞、t细胞或b细胞从血细胞中分
离。或者,可以制备预先分离的单核细胞。然而,重编程因子rna可转移到尚未根据细胞表面标记分离的血细胞。
[0374]
cd34阳性细胞是干细胞/祖细胞,并且倾向于重新编程。当使用作为cd3阳性细胞的t细胞生产ips细胞时,源自t细胞的ips细胞保留tcr重组系统,并且因此易于高效诱导分化成t细胞。
[0375]
分离cd34阳性细胞的方法如下。
[0376]
将10μl的il-6(100μg/ml)、10μl的scf(300μg/ml)、10μl的tpo(300μg/ml)、10μl的flt3配体(300μg/ml)和10μl的il-3(10μg/ml)添加至10ml的无血清培养基(stemspan h3000,stemcell technologies股份有限公司),以制备血细胞培养基(造血干/祖细胞培养基)。
[0377]
将1ml至6ml,优选2ml的血细胞培养基置于6孔板的一个孔中。为了防止培养基的蒸发,将1ml至6ml,优选2ml的pbs置于其余五个孔的每一个孔中。然后,将6孔板置于4℃至42℃,优选37℃的培养箱中并温育。
[0378]
制备含有添加至20ml的pbs的10μl至1ml,优选80μl的edta(500mmol/l)和10μl至1ml,优选200μl的fbs的柱缓冲液。将含有1
×
104至1
×
109,优选2
×
107个单核细胞的单核细胞悬浮液分配到15ml的管,并将单核细胞悬浮液在4℃至42℃,优选4℃下,以100
×
g至3000
×
g,优选300
×
g离心10分钟。在离心之后,除去上清液,并将单核细胞悬浮于100μl至1ml,优选300μl的柱缓冲液中。
[0379]
将10μl至1ml,优选100μl的fcr阻断试剂(miltenyi biotec k.k.)和10μl至1ml,优选100μl的cd34微珠试剂盒(miltenyi biotec k.k.)添加至15ml的管中的单核细胞悬浮液。fcr阻断试剂用于加强微珠标记的特异性。然后,将单核细胞悬浮液混合,并在4℃至42℃,优选4℃下静置1分钟至2小时,优选30分钟。
[0380]
接下来,通过添加1ml至15ml,优选10ml的柱缓冲液来稀释15ml的管中的单核细胞悬浮液,并将其在4℃至42℃,优选4℃下,以100
×
g至1000
×
g,优选300
×
g离心1分钟至2小时,优选10分钟。在离心之后,使用抽吸器除去15ml的管中的上清液,并通过添加10μl至10ml,优选500μl的柱缓冲液使单核细胞重新悬浮。
[0381]
将自动磁性细胞分离装置用柱(ms柱,miltenyi biotec k.k.)附接到自动磁性细胞分离装置(minimacs分离单元,miltenyi biotec k.k.),并通过添加10μl至10ml,优选500μl的柱缓冲液来洗涤该柱。接下来,将单核细胞置于柱中。进一步将10μl至10ml,优选500μl的柱缓冲液置于柱中,将柱洗涤一次至十次,优选三次。然后,将柱从自动磁性细胞分离装置上取下,并置入15ml的管中。接着,将10μl至10ml,优选1000μl的柱缓冲液置于柱中,并立即推动注射器以将cd34阳性细胞洗脱到15ml的管中。
[0382]
用trypan blue将10μl的cd34阳性细胞悬浮液染色,并用血细胞计数器对细胞数量计数。将15ml的管中的cd34阳性细胞悬浮液在4℃至42℃,优选4℃下,以100
×
g离心至1000
×
g,优选300
×
g离心1分钟至2小时,优选十分钟。在离心之后,使用抽吸器除去上清液。另外,使cd34阳性细胞重新悬浮于预先加温的血细胞培养基中,并将cd34阳性细胞接种于培养板上。然后,将cd34阳性细胞在4℃至42℃,优选37℃下,在1%至20%,优选5%的co2环境中培养6天。在该培养期间,培养基更换可为不必要的。
[0383]
基于除cd34以外的标记而分离细胞的方法与分离cd34阳性细胞的方法相同。
[0384]
重编程因子rna待转移至其的血细胞例如在t细胞培养基或造血干/祖细胞培养基中进行培养。在生产源于t细胞的ips细胞的情况下,使用t细胞培养基。在从cd34阳性细胞生产ips细胞的情况下,使用造血干/祖细胞培养基。培养条件包括例如5%的co2浓度、25%或更低的氧浓度和37℃或更低的温度。
[0385]
重编程因子rna待转移至其的血细胞使用诸如matrigel(corning股份有限公司)、cellstart(r)(thermo fisher scientific股份有限公司)或laminin511(nippi股份有限公司)的基膜基质以无饲养细胞的方式进行培养。
[0386]
可使用诸如以下培养液:primate es细胞培养基、reprostem、reproff、reproff2、reproxf(reprocell股份有限公司)、mtesr1、tesr2、tesre8、reprotesr(stemcell technologies股份有限公司)、pluristem(r)人es/ips培养基(merck kgaa)、用于人ips和es细胞的nutristem(r)xf/ff培养基、pluriton重编程培养基(stemgent股份有限公司)、pluristem(r)、stemfitak02n、stemfit ak03(ajinomoto股份有限公司)、用于hesc/ips的的无血清和无饲养细胞培养基esc-sure(r)(applied stemcell股份有限公司),以及l7(r)hpsc培养系统(lonza japan有限责任公司)。
[0387]
对于漂浮培养,将血细胞置于旋转瓶中并在搅拌下培养。或者,可将血细胞置于0.001%至10%的结冷胶溶液中(选自由脱乙酰结冷胶、透明质酸、鼠李胶、定优胶、黄原胶、角叉菜胶、岩藻依聚糖、果胶、果胶酸、果胶酯酸、硫酸乙酰肝素、肝素、硫酸肝素、硫酸角质素、硫酸软骨素、硫酸皮肤素、硫酸鼠李聚糖及其盐组成的组中的至少一种聚合物化合物),或温敏性凝胶中并培养。凝胶培养基可含有甲基纤维素。其中所含的甲基纤维素抑制细胞之间的聚集。
[0388]
温敏性凝胶可含有选自以下各项中的至少一种:聚(甘油单甲基丙烯酸酯)(pgma)、聚(2-羟丙基甲基丙烯酸酯)(phpma)、聚异丙基丙烯酰胺、聚(n-异丙基丙烯酰胺)(pnipam)、胺基封端的、羧酸封端的、马来酰亚胺封端的、n-羟基丁二酰亚胺(nhs)酯封端的、三乙氧基硅烷封端的、聚(n-异丙基丙烯酰胺-共-丙烯酰胺)、聚(n-异丙基丙烯酰胺-共-丙烯酸)、聚(n-异丙基丙烯酰胺-共-丙烯酸丁酯)、聚(n-异丙基丙烯酰胺-共-甲基丙烯酸)、聚(n-异丙基丙烯酰胺-共-甲基丙烯酸-共-十八烷基丙烯酸酯)和n-异丙基丙烯酰胺。
[0389]
培养基可含有选自由钙粘蛋白、层粘连蛋白、纤连蛋白和玻连蛋白组成的组中的至少一种物质。
[0390]
重编程因子rna转移到血细胞。重编程因子rna包括例如oct3/4mrna、sox2 mrna、klf4 mrna和c-myc mrna。重编程因子rna还可包括选自由以下各项组成的组中的至少一种因子的mrna:lin28a、lin28b、glis1、foxh1、p53-显性失活、p53-p275s、l-myc、nanog、dppa2、dppa4、dppa5、zic3、bcl-2、e-ras、tpt1、sall2、nac1、dax1、tert、znf206、foxd3、rex1、utf1、klf2、klf5、esrrb、mir-291-3p、mir-294、mir-295、nr5a1、nr5a2、tbx3、mbd3sh、th2a和th2b。这些mrna可从trilink biotechnologies股份有限公司购得。
[0391]
每种mrna可用选自由以下各项组成的组中的至少一种来修饰:假尿苷(ψ)、5-甲基尿苷(5meu)、n1-甲基假尿苷(me1ψ)、5-甲氧基尿苷(5mou)、5-羟甲基尿苷(5hmu)、5-甲酰尿苷(5fu)、5-羧甲基尿苷(5camu)、噻吩鸟苷(thg)、n4-甲基胞苷(me4c)、5-甲基胞苷(m5c)、5-甲氧基胞苷(5moc)、5-羟甲基胞苷(5hmc)、5-羟基胞苷(5hoc)、5-甲酰胞苷(5fc)、5-羧基胞苷(5cac)、n6-甲基-2-氨基腺苷(m6dap)、二氨基嘌呤(dap)、5-甲基尿苷(m5u)、
1003)和reprorna(r)转染试剂(stemcell technologies股份有限公司)可用作rna脂质转染试剂。
[0400]
用于以重编程因子rna的脂质转染的血细胞的数量为例如1至1
×
108个细胞、1
×
104个细胞至5
×
106个细胞或5
×
105个细胞至5
×
106个细胞。每毫升培养液中用于脂质转染的重编程因子rna的量为例如每次5ng至50μg、50ng至10μg或600ng至3μg。用于脂质转染的脂质转染试剂的量为例如每次0.1μl至500μl、1μl至100μl或1μl至40μl。以重编程因子的脂质转染每次进行0.1小时或更长且24小时或更短、2小时或更长且21小时或更短、12小时30分钟或更长且18小时30分钟或更短,或18小时。例如,当使用12孔板并且细胞的数量为4
×
105时,使用6μl的rnaimax或3μl的messengermax。
[0401]
重复进行用于重编程的脂质转染,例如两天一次或一天一次或五天或更长且九天或更短时间一次、六天或更长且八天或更短时间一次,或七天一次。然而,当mrna是复制rna时,可进行一次脂质转染。在以重编程因子rna的脂质转染中使用的培养基是例如低血清培养基,诸如opti-mem(gibco)。
[0402]
例如,通过使用流式细胞仪分析对于选自tra-1-60、tra-1-81、ssea-1和ssea5的至少一种表面标记是否呈阳性,确认是否从血细胞中诱导出了诱导多能干细胞或血细胞是否重编程为诱导多能干细胞,其中这些标记是表明未分化的细胞表面标记。tra-1-60是ips/es细胞特异性的抗原,并且在体细胞中未检测到。因为ips细胞仅可从tra-1-60阳性片段获得,所以tra-1-60阳性细胞视为ips细胞中的一种。
[0403]
在上述根据本发明实施例的生产诱导多能干细胞方法中,通过将允许重编程因子表达的rna转移到体细胞例如血细胞中并表达重编程因子,生产出了诱导多能干细胞。因此,有可能在不将重编程因子整合到体细胞的dna中的情况下生产出诱导多能干细胞。
[0404]
在用于生产诱导多能干细胞的常规方法中,将重编程因子插入体细胞dna中。这损伤基因组并触发细胞的肿瘤发生。相比之下,在根据本发明实施例的生产诱导多能干细胞的方法中,因为采用了编码重编程因子的rna,所以有可能在不将基因插入基因组的情况下生产诱导多能干细胞,并且不存在相关的肿瘤发生的可能性。因此,通过根据本发明实施例的生产方法生产的诱导多能干细胞使得有可能满足良好的临床可用细胞的制造实践。
[0405]
在使用逆转录病毒或慢病毒生产诱导多能干细胞的常规方法中,病毒保留在生产的诱导多能干细胞中。相比之下,在根据本发明实施例的生产诱导多能干细胞的方法中,由于通过脂质转染转移重编程因子rna,因此不需要病毒。因此,所生产的诱导多能干细胞中不存在病毒。同样,就此方面来说,通过根据本发明实施例的生产方法生产的诱导多能干细胞使得有可能满足良好的临床可用细胞的制造实践。
[0406]
使用电穿孔生产诱导多能干细胞的常规方法在诱导之前极大地损伤细胞并且破坏大量细胞。相比之下,在根据本发明实施例的生产诱导多能干细胞的方法中使用的脂质转染在诱导前对细胞几乎没有损伤并且不破坏大量细胞。另外,脂质转染不需要昂贵的装备,并且通过简便的过程进行。
[0407]
为了从成纤维细胞生产诱导多能干细胞,需要通过高创性的活组织检查采集皮肤细胞。相比之下,根据本发明实施例的生产诱导多能干细胞的方法使得有可能通过低创性采血来采集血细胞。通常,从血液采集可获得足够数量的生产诱导多能干细胞所需的血细胞。因此,与成纤维细胞不同,血细胞在诱导诱导多能干细胞之前不必增殖。此外,血细胞不
存在增殖培养期间可能发生的dna损伤的风险。此外,与皮肤细胞不同,可在不排出体外的情况下采集血细胞。因此,可在干净的密封系统中从在血液采集阶段获得的血细胞中诱导诱导多能干细胞。就此而言,血细胞也适合临床使用。
[0408]
(实例13)
[0409]
(制备)
[0410]
从健康的成年男性获得人血细胞。另外,准备经修饰的mrna(trilink biotechnologies股份有限公司)、非粘附培养皿、15ml管、50ml管、ficoll、流式细胞仪(becton,dickinson and company)、抗cd34抗体(miltenyi bioteck.k.)、抗cd3抗体(miltenyi biotec k.k.)、macs(r)缓冲液(miltenyi biotec k.k.)、t细胞培养基、低血清培养基(opti-mem,gibco)、sirna转移试剂(lipofectamine(r)rnaimax,thermo fisher science股份有限公司)和抗tra-1-60抗体(becton,dickinson and company)。
[0411]
t细胞(cd3阳性细胞)培养基是下述a培养基和b培养基的混合溶液。a培养基是15ml的x vivo-10(lonza japan有限责任公司,04-743q)和il-2(10μg/ml)的混合溶液。通过将x vivo-10和50μl的dynabeads cd3/cd28(life technologies股份有限公司,111-31d)混合到1.5ml的管中,涡旋管5秒,然后旋转管,在dynamag-2(thermo fisher science股份有限公司)上静置管,并在静置1分钟后除去上清液来制备b培养基。
[0412]
向10ml的无血清培养基(stemspan h3000,stemcell technologies股份有限公司)中添加10μl的il-6(100μg/ml)、10μl的scf(300μg/ml)、10μl的tpo(300μg/ml)、10μl的flt3配体(300μg/ml)和10μl的il-3(10μg/ml)以制备血细胞培养基(造血干/祖细胞培养基)。
[0413]
另外,制备含有oct3/4mrna的溶液、含有sox2 mrna的溶液、含有klf4 mrna的溶液、含有c-myc mrna的溶液、含有lin28a mrna的溶液和含有绿色荧光蛋白(gfp)mrna的溶液,以使其相应浓度为100ng/μl。接下来,将385μl的含有oct3/4mrna的溶液、119μl的含有sox2mrna的溶液、156μl的含有klf4 mrna的溶液、148μl的含有c-myc mrna的溶液、83μl的含有lin28a mrna的溶液和110μl的含有gfp mrna的溶液混合以获得重编程因子混合溶液。将获得的重编程因子混合溶液以50μl/管分配到1.5ml的不含rnase的管(eppendorf管,eppendorf ag)中,并储存在-80℃的冰箱中。
[0414]
(单核细胞的制备)
[0415]
将离心机设定为18℃。采集5ml至50ml的血液,并将edta添加至血液,随后温和地混合。将人淋巴细胞分离用培养基(ficoll-paque premium,ge healthcare japan股份有限公司)以5ml/管分配到两个15ml的管。向血液中添加5ml的pbs进行稀释,并将5ml的稀释后的血液层积在各管中的人淋巴细胞分离用培养基上。此时,将稀释后的血液缓慢地添加至培养基上,使得血液沿着管壁向下流动,以不干扰分界面。
[0416]
将管中的溶液在18℃下以400
×
g离心30分钟。在这一操作中,加速和减速都缓慢地进行。在离心之后,管中出现白色混浊的中间层。该白色混浊中间层含有单核细胞。用移液管逐渐回收管中的白色混浊的中间层,并将其转移到新15ml的管中。在这一操作中,注意避免吸出下层。可从一个管回收约1ml的量的白色混浊的中间层。将来自两个管的中间层一起转移到一个管。
[0417]
向回收的单核细胞添加12ml的pbs,并进一步将溶液在18℃下以200
×
g离心10分
钟。然后,使用抽吸器吸去溶液的上清液,并通过添加3ml的具有已知组成的无血清造血细胞培养基(x-vivo(r)10,lonza japan有限责任公司)使单核细胞悬浮,以获得单核细胞悬浮液。将单核细胞悬浮液的10μl等分试样用trypan blue染色并使用血细胞计数器计数。
[0418]
(分离cd34或cd3阳性细胞)
[0419]
使1
×
107个单核细胞在100μl的4℃的溶液中与抗cd34的抗体或抗cd3的抗体反应15分钟。在反应之后,向溶液添加5ml的macs(r)缓冲液(miltenyi biotec k.k.),并将混合溶液以270g离心。在离心之后,除去上清液,并向细胞添加1ml的macs缓冲液。然后,使用自动磁性细胞分离装置(automacs,miltenyi biotec k.k.)的分离程序分离单核细胞中的cd34阳性细胞或cd3阳性细胞。
[0420]
(培养分离的细胞)
[0421]
将5
×
106个分离的单核细胞悬浮在1ml的t细胞培养基或造血干/祖细胞培养基中,接种到12孔板中并培养。培养条件包括5%的co2浓度、19%的氧气浓度和37℃的温度。
[0422]
(重编程因子的脂质转染)
[0423]
将100μl的低血清培养基(opti-mem(r),gibco)和25μl的重编程因子混合溶液混合以制备第一混合溶液。此外,将112.5μl的低血清培养基(opti-mem(r),gibco)和12.5μl的sirna转移试剂(lipofectamine(r)rnaimax,thermo fisher science股份有限公司)混合以制备第二混合溶液。接着,混合第一混合溶液和第二混合溶液,在室温下静置15分钟,以制备脂质转染反应溶液。
[0424]
在培养过程中,将获得的脂质转染反应溶液以60μl/孔缓慢添加至含有单核细胞的12孔板,随后在37℃下以无饲养细胞方式培养单核细胞18小时。
[0425]
培养条件包括5%的co2浓度、19%的氧气浓度和37℃的温度。当添加脂质转染反应溶液时,单核细胞的密度是3
×
106个细胞。18小时之后,将单核细胞回收到15ml的管中并以300g离心,随后除去上清液。然后,将1.25ml的用于cd34的血细胞培养基添加至15ml的管。将单核细胞悬浮液放回与上述相同的12孔板。将单核细胞在37℃下以无饲养细胞的方式培养过夜。
[0426]
培养条件包括5%的co2浓度和19%的氧气浓度。这些步骤每两天重复,持续七天。
[0427]
(确认gfp表达)
[0428]
在脂质转染开始后的第7天,总共四次脂质转染的细胞的密度是3
×
106个细胞。从12孔板中取出等分试样,并在荧光显微镜下确认gfp的表达。因此,如图35所示,确认gfp的表达。根据这些结果,确认蛋白质是从转染有mrna的单核细胞所携带的mrna合成。
[0429]
(确认tra-1-60表达)
[0430]
在脂质转染开始后的第7天,从12孔板中取出细胞的等分试样,并用抗tra-1-60(在其中已经开始重编程的细胞上特异性表达的表面抗原)的别藻蓝蛋白(apc)荧光染料标记的抗体将这样取出的细胞染色。然后,使用荧光激活细胞分选仪(facs(r),becton,dickinson and company)确认tra-1-60-阳性细胞的百分比,以确认在细胞中开始重编程使得ips细胞基因表达以生成ips细胞。
[0431]
如图36所示,以x轴上的自发荧光强度相对于y轴上的荧光标记的抗-tra-1-60抗体的荧光强度制作点图。在不携带基因的阴性对照中未检测到tra-1-60阳性细胞。相反,在实验1、2、3中检测到tra-1-60阳性细胞。实验1描述了从基于标记未分离的全部单核细胞诱
导ips细胞的结果。实验2描述了从cd3阳性分离的细胞诱导ips细胞的结果。实验3描述了从cd34阳性分离的细胞诱导ips细胞的结果。这些结果表明,有可能通过使用以重编程因子rna的脂质转染将重编程因子转移到血源细胞中来诱导ips细胞。
[0432]
(第六实施例)
[0433]
根据本发明实施例的从动物细胞生产体细胞的方法包括:制备动物细胞;和通过脂质转染将诱导物核糖核酸(rna)转移到动物细胞中以将动物细胞分化成体细胞。
[0434]
动物细胞包括干细胞。诱导多能干细胞(ips细胞)和胚胎干细胞(es细胞)均可用作干细胞。动物细胞可为人成纤维细胞或人血细胞。
[0435]
可使用诸如primate es细胞培养基、mtesr1、tesr2或tesre8(stemcell technologies股份有限公司)的培养液来培养干细胞。
[0436]
用于培养干细胞的培养基可含有凝胶。凝胶可含有选自由脱酰结冷胶、结冷胶、透明质酸、鼠李胶、定优胶、黄原胶、角叉菜胶、岩藻依聚糖、果胶、果胶酸、果胶酯酸、硫酸乙酰肝素、肝素、硫酸肝素、硫酸角质素、硫酸软骨素、硫酸皮肤素、硫酸鼠李聚糖及其盐组成的组的至少一种聚合物化合物。凝胶培养基可含有甲基纤维素。其中所含的甲基纤维素抑制细胞之间的聚集。
[0437]
凝胶可为温敏性凝胶。温敏性凝胶可为选自以下各项中的至少一种:聚(甘油单甲基丙烯酸酯)(pgma)、聚(2-羟丙基甲基丙烯酸酯)(phpma)、聚(n-异丙基丙烯酰胺)(pnipam)、胺基封端的、羧酸封端的、马来酰亚胺封端的、n-羟基丁二酰亚胺(nhs)酯封端的、三乙氧基硅烷封端的、聚(n-异丙基丙烯酰胺-共-丙烯酰胺)、聚(n-异丙基丙烯酰胺-共-丙烯酸)、聚(n-异丙基丙烯酰胺-共-丙烯酸丁酯)、聚(n-异丙基丙烯酰胺-共-甲基丙烯酸)、聚(n-异丙基丙烯酰胺-共-甲基丙烯酸-共-十八烷基丙烯酸酯)和n-异丙基丙烯酰胺。
[0438]
用于培养干细胞的培养基可含有选自由钙粘蛋白、层粘连蛋白、纤连蛋白和玻连蛋白组成的组中的至少一种物质。
[0439]
从动物细胞生产的体细胞是例如神经元细胞,但体细胞不限于此。例如,可生产诸如心肌细胞、肝细胞、视网膜细胞、角膜细胞和血细胞的体细胞。在生产神经元细胞的情况下,待转移到动物细胞中的诱导物rna包括例如神经原素2(ngn2)mrna。ngn2是分化成神经元细胞所必需的转换蛋白。诱导物rna可包含对应于药物抗性基因的mrna。该药物是例如抗生素,诸如嘌呤霉素、新霉素、杀稻瘟菌素、g418、潮霉素或博莱霉素。携带诱导物rna的细胞表现出药物抗性。
[0440]
包含在诱导物rna中的每种mrna可用选自由以下各项组成的组中的至少一种来修饰:假尿苷(ψ)、5-甲基尿苷(5meu)、n1-甲基假尿苷(me1ψ)、5-甲氧基尿苷(5mou)、5-羟甲基尿苷(5hmu)、5-甲酰尿苷(5fu)、5-羧甲基尿苷(5camu)、噻吩鸟苷(thg)、n4-甲基胞苷(me4c)、5-甲基胞苷(m5c)、5-甲氧基胞苷(5moc)、5-羟甲基胞苷(5hmc)、5-羟基胞苷(5hoc)、5-甲酰胞苷(5fc)、5-羧基胞苷(5cac)、n6-甲基-2-氨基腺苷(m6dap)、二氨基嘌呤(dap)、5-甲基尿苷(m5u)、2'-o-甲基尿苷(um或m2'-ou)、2-硫代尿苷(s2u)和n6-甲基腺苷(m6a)。
[0441]
mrna可进行多聚腺苷酸化。mrna可通过体外转录(ivt)rna的多聚腺苷酸化来制备。通过使用编码聚(a)尾的dna模板,mrna在ivt期间可进行多聚腺苷酸化。mrna可加帽。为了最大化细胞中的表达效率,绝大多数mrna分子可含有帽。
[0442]
mrna可具有5'帽[m7g(5')ppp(5')g]结构。该序列稳定mrna并促进mrna转录。从具有5'三磷酸的mrna,可通过去磷酸化处理来除去5'三磷酸。mrna可具有[3'o-me-m7g(5')ppp(5')g]作为抗逆转录帽类似物(arca)。arca是插入转录起点上游的序列,并且使mrna转录效率翻倍。mrna可具有聚(a)尾。
[0443]
诱导物rna包括例如ngn2-t2a-puro mrna(trilink biotechnologies股份有限公司,对应于seq id no:1中描述的dna的rna)。用ngn2-t2a-puro mrna(trilink biotechnologies股份有限公司)转染的细胞生产神经原素2(ngn2),并且还表现出嘌呤霉素抗性。mrna可用抗逆转录帽类似物(arca)加帽,多聚腺苷酸化,并用5-甲基胞苷和假尿苷取代。5-甲基胞苷和假尿苷降低了抗体识别mrna的能力。可使用对应于seq id no:2中描述的dna的rna。seq id no:2中描述的dna是通过除去xbal限制性位点而从seq id no:1的dna衍生的。
[0444]
通过脂质转染法将诱导物rna转移到动物细胞中。脂质转染法是一种涉及通过电相互作用形成核酸(带负电的物质)和带正电的脂质的复合物,并通过胞吞或膜融合使该复合物进入细胞的方法。脂质转染法具有诸如细胞损伤小、转移效率优异、操作方便、持续时间短的优点。
[0445]
例如,lipofectamine messengermax(r)用作诱导物rna的脂质转染中的脂质转染试剂。或者,例如,lipofectamine(r)rnaimax(thermo fisher scientific股份有限公司)、lipofectamine(r)2000、lipofectamine(r)3000、neon转染系统(thermo fisher scientific股份有限公司)、stemfect rna转染试剂(stemgent)、nextfect(r)rna转染试剂(bioo scientific股份有限公司)、amaxa(r)人体t细胞nucleofector(r)试剂盒(lonza japan有限责任公司,vapa-1002)、amaxa(r)人体cd34细胞nucleofector(r)试剂盒(lonza japan有限责任公司,vapa-1003)和reprorna(r)转染试剂(stemcell technologies股份有限公司)可用作脂质转染试剂。
[0446]
在使用例如12孔板的情况下,用于以诱导物rna的脂质转染的细胞的数量为每孔1
×
104至1
×
108、5
×
104至1
×
106,或1
×
105至5
×
105。一个孔的底面积为4cm2。用于以诱导物rna的脂质转染的诱导物rna的量为每次200ng至5000ng、400ng至2000ng或500ng至1000ng。用于以诱导物rna的脂质转染的脂质转染试剂的量为0.1μl至100μl、1μl至50μl或1.5μl至10μl。
[0447]
用于以诱导物rna的脂质转染的培养基是例如低血清培养基,诸如opti-mem(r)(gibco)。用于诱导物rna的脂质转染期间以及这种脂质转染之前或之后的培养基可含有b18r蛋白。b18r蛋白减轻细胞的先天性抗病毒应答。b18r蛋白可用于抑制与rna插入细胞相关的免疫应答所导致的细胞死亡。然而,因为根据本实施例的从动物细胞生产体细胞的方法在短时间内使动物细胞分化成体细胞,所以培养基可不含b18r蛋白或可含有稀释浓度为0.01%至1%的b18r蛋白。
[0448]
自已诱导物rna的脂质转染后的十天、九天、八天或七天内,动物细胞分化成体细胞。当待生产的体细胞是神经元细胞时,根据它们对ngn2、β-iii微管蛋白、map2、psa-ncam或vglu是否呈阳性来确认它们是否分化成神经元细胞。ngn2是分化成神经元细胞所必需的转换蛋白。β-iii微管蛋白、map2、psa-ncam和vglu是标记神经元细胞的标记,并且是神经突中微管的组成蛋白。
[0449]
当诱导物rna包含对应于药物抗性基因的mrna时,可在脂质转染之后选择表现出药物抗性的细胞。当诱导物rna包含例如对应于嘌呤霉素抗性基因的mrna时,通过将脂质转染的细胞暴露于嘌呤霉素以选择携带诱导物rna的细胞可破坏除携带诱导物rna的细胞以外的细胞。诱导物rna可包含对应于选自新霉素、杀稻瘟菌素、g418、潮霉素、博莱霉素等的任何抗生素的基因的mrna作为对应于药物抗性基因的mrna。
[0450]
根据上述本发明实施例的用于从动物细胞生产体细胞的方法有可能通过在包括ips/es细胞等动物细胞中表达编码特定基因的rna而高效地生产诸如神经元细胞的体细胞而损伤包括ips/es细胞等动物细胞中的基因。
[0451]
用激素或化学物质从ips/es细胞等生产体细胞的方法需要非常长的时间来生产体细胞。相比之下,根据本发明实施例的从动物细胞生产体细胞的方法有可能在很短的时间内生产体细胞。
[0452]
在使用激素或化学物质从包括ips/es细胞等动物细胞生产体细胞的方法中,仅包括ips/es细胞等动物细胞中的一些细胞转化为感兴趣的体细胞。相比之下,根据本发明实施例的从动物细胞生产体细胞的方法通过rna转移将90%或更多的细胞转化为感兴趣的体细胞。
[0453]
在使用激素或化学物质从ips/es细胞等生产体细胞的方法中,即使使用相同的方案,其导致集落之间的差异,使得一些集落变成感兴趣的体细胞而其他集落不变。相比之下,根据本发明实施例的从动物细胞生产体细胞的方法有可能实现多个集落的高效率诱导分化。
[0454]
在使用细胞因子等诱导诸如es/ips细胞的未分化细胞群的分化来生产移植用细胞的情况下,有可能未分化细胞残留在移植用细胞内。此类残留未分化细胞具有由于其自身在移植部位的细胞分裂和增殖而形成畸胎瘤等的风险。相比之下,根据本发明实施例的从动物细胞生产体细胞的方法有可能基于药物来选择携带诱导物rna的细胞,因为药物抗性基因可与其共同表达。因此,通过本发明的方法生产的细胞可避免未分化细胞污染和畸胎瘤形成等的风险,并且因此适于医学移植。
[0455]
在使用脂质转染法的本发明实施例的从动物细胞生产体细胞的方法中,不使用病毒。因此,干细胞的基因不被损伤,并且生产的体细胞没有相关的肿瘤发生风险,可以用于临床治疗。
[0456]
使用病毒从干细胞生产体细胞的方法需要大肠杆菌来生产和增殖病毒载体。然而,通过转移使用非人类生物生产的物质来生产的细胞不适合临床应用。相比之下,根据本发明实施例的从动物细胞生成体细胞的方法可通过使用脂质转染法将rna转移到包括ips/es细胞等动物细胞中。因为rna是化学物质并且可人工合成,所以rna可在不使用诸如大肠杆菌的生物体的情况下制造并且适合临床应用。
[0457]
例如,ips细胞在完全密封系统的清洁环境中从血细胞生产,随后体细胞在完全密封系统的清洁环境中从ips细胞生产。在这种情况下,有可能生产更清洁和更安全的体细胞。
[0458]
此外,根据本发明实施例的从动物细胞生产体细胞的方法有可能在短时间内生产体细胞。因此,例如,不必使用抑制与mrna插入相关的免疫应答所导致的细胞死亡的b18r。即使使用这样的物质,其极稀浓度是可能的。
[0459]
(实施例14)
[0460]
制备包覆有可溶性基膜制剂(matrigel,corning股份有限公司)的12孔培养皿。将含有浓度为10umol/l的rock(rho相关的卷曲螺旋形成的激酶/rho结合激酶)抑制剂(selleck chemicals)的无饲养细胞培养基(mtesr(r)1,stemcell technologies股份有限公司)置于每个孔中。rock抑制剂抑制细胞死亡。
[0461]
将ips细胞分散在用于组织/培养细胞的脱离/分离/分散溶液(accutase,innovative cell technologies股份有限公司)中,并接种到12孔培养皿中。将待转染的细胞以每孔4
×
105个细胞的密度接种。一个孔的底面积为4cm2。将未转染的对照细胞以每孔2
×
105个细胞的密度接种。然后,将细胞在无饲养细胞培养基中培养24小时。在该培养中,温度为37℃,co2浓度为5%,氧浓度为25%或更低。
[0462]
将1.25ml的无异源培养基(pluriton,stemgent股份有限公司)、0.5μl的pluriton补充物(stemgent股份有限公司)和2μl的含有浓度为100ng/μl的b18r重组蛋白的溶液(ebioscience)混合以制备转染培养基。在转染之前,将每孔中的无饲养细胞培养基更换为转染培养基,细胞在其中在37℃下培养2小时。
[0463]
制备绿色荧光蛋白(gfp)mrna(trilink biotechnologies股份有限公司)。将mrna用抗逆转录帽类似物(arca)加帽,多聚腺苷酸化,并用5-甲基胞苷和假尿苷取代。
[0464]
分别准备1.5ml的微量离心分离管a和1.5ml的微量离心分离管b,以对应于孔数。
[0465]
将62.5μl的低血清培养基(opti-mem,gibco)置于各管a中,然后向其添加1.875μl的用于mrna转移的试剂(lipofectamine messengermax,invitrogen股份有限公司)并充分混合以制备第一反应溶液。然后,将管a在室温下轻敲10分钟,使得第一反应溶液混合。
[0466]
将62.5μl的低血清培养基(opti-mem(r),gibco)置于各管b中,向其添加500ng的gfp mrna(trilink biotechnologies股份有限公司)并充分混合以制备第二反应溶液。
[0467]
将第二反应溶液添加至管a中的第一反应溶液以制备混合反应溶液。然后,将管a在室温下轻敲5分钟,使得脂质体形成。接下来,将混合反应溶液添加至每个孔,并在37℃下静置过夜。因此,向每孔添加500ng的gfp mrna。
[0468]
第二天,在荧光显微镜下观察细胞。因此,如图37和图38所示,使用messengermax转染的细胞最强烈地产生了颜色。如图39所示,使用messengermax转染的细胞也表现出最高的存活率。这揭示了messengermax最适合mrna转移。这些结果表明,有可能通过使用脂质转染试剂和rna进行mrna转移来在ips细胞中表达蛋白质。
[0469]
(实例15)
[0470]
制备包覆有可溶性基膜制剂(matrigel,corning股份有限公司)的12孔培养皿。将含有浓度为10μmol/l的rock(rho相关的卷曲螺旋形成的激酶/rho结合激酶)抑制剂(selleck chemicals)的无饲养细胞培养基(mtesr(r)1,stemcell technologies股份有限公司)置于每个孔中。rock抑制剂抑制细胞死亡。
[0471]
将ips细胞分散在用于组织/培养细胞的脱离/分离/分散溶液(accutase,innovative cell technologies股份有限公司)中,并接种到12孔培养皿中。将待转染的细胞以每孔4
×
105个细胞的密度接种。将未转染的对照细胞以每孔2
×
105个细胞的密度接种。然后,将细胞在无饲养细胞培养基中培养24小时。
[0472]
将1.25ml的无异源培养基(pluriton,stemgent股份有限公司)、0.5μl的pluriton
补充物(stemgent股份有限公司)和2μl的含有浓度为100ng/μl的b18r重组蛋白的溶液(ebioscience)混合以制备转染培养基。在转染之前,将每孔的无饲养细胞培养基更换成转染培养基,细胞在其中在37℃下培养2小时。
[0473]
制备ngn2-t2a-puro mrna(trilink biotechnologies股份有限公司)和绿色荧光蛋白(gfp)mrna(trilink biotechnologies股份有限公司)。每种mrna用抗逆转录帽类似物(arca)加帽,多聚腺苷酸化,并用5-甲基胞苷和假尿苷取代。此外,通过二氧化硅膜纯化mrna,并将其与用于mrna转移的试剂(lipofectamine messengermax,invitrogen股份有限公司)一起制备成含有1mmol/l的柠檬酸钠(ph6)溶液(作为溶剂)。分别准备1.5ml的微量离心分离管a和1.5ml的微量离心分离管b,以对应于孔数。
[0474]
将62.5μl的低血清培养基(opti-mem(r),gibco)置于各管a中,然后向其添加1.875μl的用于mrna转移的试剂(lipofectamine messengermax(r),invitrogen股份有限公司)并充分混合以制备第一反应溶液。然后,将管a在室温下轻敲10分钟,使得第一反应溶液混合。
[0475]
将62.5μl的低血清培养基(opti-mem(r),gibco)置于各管b中,然后向其添加500ng的ngn2-t2a-puro mrna(trilink biotechnologies股份有限公司)和1500ng的gfp mrna(trilink biotechnologies股份有限公司)并充分混合以制备第二反应溶液。
[0476]
将第二反应溶液添加至管a中的第一反应溶液以制备混合反应溶液。然后,将管a在室温下轻敲5分钟,使得脂质体形成。接下来,将混合反应溶液添加至每个孔,并在37℃下静置过夜。因此,向每孔添加了500ng的ngn2mrna和100ng的gfp mrna。
[0477]
如图40所示,通过观察mrna转移一天后的细胞,使用messengermax转染的细胞最强烈地产生了颜色。
[0478]
然后,持续两天每天用含有浓度为10μmol/l的rock抑制剂(selleck chemicals)和浓度为1mg/l的抗生素(嘌呤霉素)的神经分化培养基(n2/dmem/f12/neaa,invitrogen股份有限公司)完全更换培养基,以选择mrna转染的细胞。在第3天,用含有浓度为200ng/ml的b18r重组蛋白的溶液(ebioscience)的神经分化培养基(n2/dmem/f12/neaa,invitrogen股份有限公司)更换培养基。然后,每次用与以上相同的培养基将培养基更换一半,直到第7天。
[0479]
在第7天,从每个孔取出培养基,并用1ml的pbs洗涤孔。然后,将4%的pfa置入其中,并使其在4℃下与细胞反应15分钟以进行固定。然后,在用pbs洗涤2次后,将各一级抗体用含有5%的ccs和0.1%的triton的pbs溶液稀释,并以500μl/孔添加。使用的一级抗体是兔抗人tuj1抗体(biolegend 845501)和小鼠抗大鼠和人ngn2抗体(r&d systems股份有限公司)。用缓冲液将兔抗人tuj1抗体(biolegend 845501)1/1000稀释,或者用缓冲液将小鼠抗大鼠和人ngn2抗体(r&d systems股份有限公司)1/75稀释,并用缓冲液将dapi 1/10000稀释。将这些稀释液添加至每个孔并使其在室温下反应1小时。抗tuj1的抗体是抗β-ιιι微管蛋白的抗体。
[0480]
在室温下反应1小时之后,向各孔添加1ml的pbs,并在孔中充分扩散,随后弃去pbs。再次向其添加pbs,然后弃去。以500μl/孔添加含有1/1000稀释的驴抗小鼠igg(h+l)二级抗体-alexa fluor(r)555复合物(thermo fisher scientific股份有限公司)或1/1000稀释的驴抗兔igg(h+l)二级抗体-alexa fluor(r)647复合物(thermo fisher scientific
股份有限公司)的含有二级抗体的透化作用缓冲液,并使其在室温下反应30分钟。
[0481]
在室温下反应30分钟之后,用pbs洗涤细胞两次,并在荧光显微镜下观察以对发荧光的细胞进行计数。
[0482]
图41是在荧光显微镜下观察时拍摄的细胞的照片,其中细胞在通过脂质转染转移ngn2-t2a-puro mrna并随后添加嘌呤霉素之后培养2天,进一步在不添加嘌呤霉素的情况下培养5天,并用tujl染色。图42示出了通过上述程序使用每种转染试剂以ngn2-t2a-puro mrna转染的细胞之中在第7天的tuj-1阳性细胞的百分比。发现messengermax比rnaimax或stemfect将ips细胞转化为神经元细胞的能力高四倍或更多。
[0483]
图43示出了在荧光显微镜下观察时拍摄的细胞的照片,其中细胞在通过三重脂质转染转移ngn2-t2a-puro mrna并随后添加嘌呤霉素之后培养6天,进一步在不添加嘌呤霉素的情况下培养16天,并用map2(sigma cat#m4403)和vglut(synaptic systems cat#135302)染色。
[0484]
(实例16)
[0485]
制备包覆有可溶性基膜制剂(matrigel,corning股份有限公司)的12孔培养皿。将含有浓度为10μmol的rock(rho相关的卷曲螺旋形成的激酶/rho结合激酶)抑制剂(selleck chemicals)的无饲养细胞培养基(mtesr(r)1,stemcell technologies股份有限公司)置于每个孔中。
[0486]
将ips细胞分散在用于组织/培养细胞的脱离/分离/分散溶液(accutase,innovative cell technologies股份有限公司)中,并接种到12孔培养皿中。将待转染的细胞以每孔4
×
105个细胞的密度接种。将未转染的对照细胞以每孔1
×
105个细胞的密度接种。然后,将细胞在无饲养细胞培养基中培养24小时。在该培养中,温度为37℃,co2浓度为5%,氧浓度为25%或更低。
[0487]
将1.25ml的无异源培养基(pluriton,stemgent股份有限公司)、0.5μl的pluriton补充物(stemgent股份有限公司)和2μl的含有浓度为100ng/μl的b18r重组蛋白的溶液(ebioscience)混合以制备具有b18r的转染培养基。此外,将1.25ml的无异源培养基(pluriton,stemgent股份有限公司)和0.5μl的pluriton补充物(stemgent股份有限公司)混合以制备不含b18r的转染培养基。
[0488]
在转染之前,将每孔中的无饲养细胞培养基更换为含有b18r的转染培养基或不含b18r的转染培养基,细胞在其中在37℃下培养2小时。
[0489]
制备ngn2-t2a-puro mrna(trilink biotechnologies股份有限公司)和gfp mrna(trilink biotechnologies股份有限公司)。将mrna用抗反转帽类似物(arca)加帽,多聚腺苷酸化,并用5-甲基胞苷和假尿苷取代。
[0490]
分别准备1.5ml的微量离心分离管a和1.5ml的微量离心分离管b,以对应于孔数。
[0491]
将62.5μl的低血清培养基(opti-mem,gibco)置于各管a中,向其添加1.875μl的用于mrna转移的试剂(lipofectamine messengermax,invitrogen股份有限公司),并充分混合以制备第一反应溶液。然后,将管a在室温下轻敲10分钟,使得第一反应溶液混合。
[0492]
将62.5μl的低血清培养基(opti-mem(r),gibco)置于各管b中,然后向其添加500ng的ngn2-t2a-puro mrna(trilink biotechnologies股份有限公司)和100ng的gfp mrna(trilink biotechnologies股份有限公司),并充分混合以制备第二反应溶液。
[0493]
将第二反应溶液添加至管a中的第一反应溶液中以制备混合反应溶液。然后,将管a在室温下轻敲5分钟,使得脂质体形成。接下来,将混合反应溶液添加至每个孔,并在37℃下静置过夜。因此,向每孔添加了500ng的ngn2 mrna和100ng的gfp mrna。如图44所示,制备单重转染的样品、双重转染的样品和三重转染的样品。
[0494]
然后,持续两天每天用含有浓度为10μmol/l的rock抑制剂(selleck chemicals)和浓度为1mg/l的抗生素(嘌呤霉素)的神经分化培养基(n2/dmem/f12/neaa,invitrogen股份有限公司)完全更换培养基,以选择mrna转染的细胞。在第3天,用含有浓度为200ng/ml的b18r重组蛋白的溶液(ebioscience)的神经分化培养基(n2/dmem/f12/neaa,invitrogen股份有限公司)更换培养基。然后,每次用与以上相同的培养基将培养基更换一半,直到第7天。
[0495]
在第7天,从每个孔中取出培养基,并用1ml的pbs洗涤孔。然后,将4%的pfa置入其中,并使其在4℃下与细胞反应15分钟以进行固定。然后,在用pbs洗涤2次之后,将各一级抗体用含有pbs中5%的ccs和0.1%的triton x的透化作用缓冲液稀释,并使其在室温下反应1小时。使用的一级抗体是用透化作用缓冲液1/1000稀释的小鼠抗人体tuj1抗体(biolegend845501)和用透化作用缓冲液1/150稀释的小鼠抗人体ngn2抗体(r&dsystems股份有限公司,mab3314-sp),并进一步以1/10000向其添加dapi。
[0496]
1小时之后,向各孔添加1ml的pbs,并在孔中充分扩散,随后弃去pbs。再次向其添加pbs,然后弃去。以500μl/孔添加含有1:1000稀释的驴抗小鼠igg(h+l)二级抗体-alexa fluor(r)555复合物(thermo fisher scientific股份有限公司,a-21428)或1:1000稀释的驴抗兔igg(h+l)二级抗体-alexa fluor(r)647复合物(thermo fisher scientific股份有限公司,a31573)的含有二级抗体的透化作用缓冲液,并使其在室温下反应30分钟。
[0497]
用pbs洗涤细胞两次,并在荧光显微镜下观察对发荧光的细胞进行计数。因此,如图45所示,用mrna单重转染的细胞在第9天几乎不表达gfp。另一方面,用mrna三重转染的细胞即使在第9天表达gfp。这揭示了mrna在细胞中被分解,并且蛋白质表达是短暂的。图46示出了用表达gfp的mrna三重转染的细胞在第7天的放大图像。
[0498]
上述结果表明,有可能在ips细胞接种后以rna转染几天之后诱导神经元细胞。结果还表明,因为可在短时间内诱导神经元细胞,所以培养基不必含有b18r蛋白,b18r蛋白通常用于抑制与rna插入细胞相关的免疫应答所导致的细胞死亡。
[0499]
[附图标记列表]
[0500]
10:分离装置
[0501]
20:转移前细胞溶液发送通道
[0502]
21:诱导物溶液发送机构
[0503]
30:诱导物转移装置
[0504]
31:转移后细胞溶液发送通道
[0505]
40:细胞批量生产装置
[0506]
50:重编程培养装置
[0507]
51:细胞簇溶液发送通道
[0508]
60:分割机构
[0509]
70:扩增培养装置
[0510]
71:扩增培养溶液发送通道
[0511]
72:细胞簇溶液发送通道
[0512]
80:分割机构
[0513]
90:细胞簇输送机构
[0514]
91:预包装细胞通道
[0515]
100:包装装置
[0516]
110:冷冻保存液发送机构
[0517]
200:容器
[0518]
序列表
[0519]
[0520]
[0521]
[0522]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1