一种布立西坦的制备方法与流程
一种布立西坦的制备方法
1.本技术要求享有2022年3月29日向中国国家知识产权局提交的申请号为202210316462.9,名称为“一种布立西坦的制备方法”的发明专利申请的优先权。该申请的全文以引用的方式并入本文。
技术领域
2.本发明涉及一种布立西坦的制备方法。
背景技术:
3.癫痫是神经系统的常见病,在人群中的发病率为0.6%~1.1%,其中60%~70%的患者在服用抗痫药物时仍会发作,导致一部分患者自行停止药物治疗。目前我国有约600万以上的癫痫患者,每年新发癫痫患者65万~70万,大约25%为难治性癫痫。虽然目前癫痫的诊疗取得了很大的进展,但难治性癫痫患者的数量却在日益增多。广义难治性癫痫是指使用目前的抗癫痫药物(aeds)规范治疗,不能终止其发作或已被临床证实是难治的癫痫及癫痫综合征。
4.布立西坦i是西坦类衍生物,具有广泛的抗癫痫活性和较高的安全性,该药可通过与突触囊泡蛋白2a(sv2a)结合而发挥抗癫痫作用。比利时制药巨头优时比(ucb)公布的癫痫药物布立西坦一项为期12周的iii期研究的数据表明,布立西坦可显著降低部分性发作频率并改善反应率。研究中布立西坦耐受性与既往研究一致。优时比于2015年分别向fda和欧洲药品管理局(ema)提交布立西坦的新药申请和上市许可申请并已经获批,布立西坦成为ucb旗帜性癫痫专营权中的第3个上市产品,该公司正在开展后期研究,寻求批准该药用于儿科患者,同时作为成人患者的单药疗法。布立西坦的主要不良事件的发生率与安慰剂组的发生率相似,均为轻度至中度的疲劳、头痛、鼻咽炎、恶心、嗜睡和头晕。无患者因不良事件而中断治疗。结果表明布立西坦片在辅助治疗年龄为16~65岁的难治性癫痫部分性发作患者中是有效的且耐受性良好。总体而言,布立西坦是继左乙拉西坦之后的一个前景十分良好的第三代抗癫痫类药物。
5.布立西坦i可以由多种方式制备得到,如文献journal of medical chemistry,2004,47(3),530-549和专利文献wo2005028435、wo2007031263、wo2007065634等,通常合成路线为五到十多步,但是最后均需要进行手性高压液相制备柱拆分,收率低、损失大、成本高、无法大规模生产。
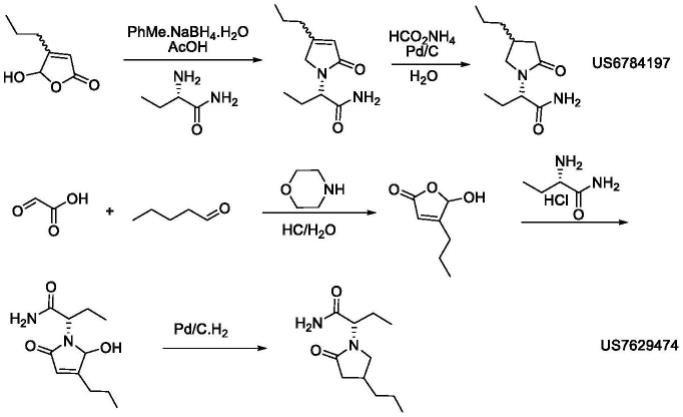
6.国外相关专利报道不多,us 6784197和us 7629474报道的合成路线如下:
[0007][0008]
但是,此路线需要使用金属钯作为催化剂进行加氢还原,对生产设备要求高,且是最后一步使用重金属,易导致成品的重金属残留,且在最后需要高效液相手性分离,收率不足20%,不利工业化生产。
[0009]
专利us 8957226、us 8338621和cn101263113b公开了一条布立西坦的制备路线。该路线以2-己烯酸乙酯为起始物料,经迈克加成得到3-硝基甲基己酸乙酯,经氢化关环得到消旋的4-正丙基吡咯烷酮,经手性制备色谱分离得到光学纯的(r)-4-正丙基吡咯烷酮,后再与2-溴丁酸甲酯反应得到(2s)-2-(2-氧代-4-正丙基-1-吡咯烷基)丁酸甲酯,再经氨解得到部分消旋化的布立西坦,最终也通过制备色谱分离得到高纯度的布立西坦。反应路线如下:
[0010][0011]
此路线使用重金属镍,且在合成中没有构建手性中心,需要手性柱进行色谱制备,
路线需要拆分收率仅10%左右,生产成本极高,不适合工业化大生产。
[0012]
kenda等(journal of medicinal chemistry,2004,47,530)报道了如下合成路线:
[0013][0014]
路线需要手性拆分(收率会远低于50%),且用到硼氢化钠还原,工业中使用硼氢化钠对工人素质,及工厂应急能力,设备均有苛刻的要求,不适合工业生产。
[0015]
专利cn105646319公开了一条布立西坦的制备路线,该路线以丙二酸二苯酯为起始物料,与(r)-环氧氯丙烷反应得到2-氧代-3-氧杂双环[3.1.0]己烷-1-甲酸苯酯,后在碘化亚铜催化下,与乙基溴化镁反应得到2-氧代-4-丙基-四氢呋喃-3-甲酸苯酯,经高温脱羧得到(r)-4-丙基-二氢呋喃-2-酮,后经三甲基溴硅烷开环,与甲醇成酯后得到(r)-3-溴代甲基己酸甲酯,最终在高温条件下与(s)-2-氨基丁酰胺缩合得到布立西坦。此路线反应收率低,摩尔收率仅为27%。反应路线如下:
[0016][0017]
因此,寻找一种收率高、反应步骤短、后处理简单、制得的产品纯度高、生产成本低、适合于工业化生产的布立西坦i的合成方法是目前急需解决的技术问题。
技术实现要素:
[0018]
本发明所要解决的技术问题是为了克服现有技术中布立西坦的合成方法反应步骤长、需要使用重金属、总收率低、后处理步骤繁琐、制得的产品纯度低、生产成本高、不适合于工业化生产等缺陷而提供了一种布立西坦的制备方法。本发明采用一锅法制备布立西
坦,反应步骤短、反应总收率高、操作简单安全、后处理步骤简单、制得的产品纯度高、生产成本低、适合于工业化生产。
[0019]
本发明提供了一种布立西坦i的制备方法,其包括以下步骤:有机溶剂中,将化合物iii与s-2氨基丁酰胺进行缩合反应,然后再在碱性条件下进行亲核取代反应,得到布立西坦i即可;
[0020][0021]
本发明所述的布立西坦i的制备方法可以在无水条件下进行。所述的无水条件可以通过控溶剂水分实现;所述的溶剂的含水量优选小于0.5%,进一步优选为小于0.1%;所述的含水量是指水的质量与反应体系中溶剂总质量的百分比。
[0022]
本发明所述的布立西坦i的制备方法可以在惰性气体保护下进行;所述的惰性气体优选氮气和/或氩气。
[0023]
本发明所述的布立西坦i的制备方法,一锅法进行可以不用分离提纯中间体ii直接制备布立西坦i。
[0024]
本发明中,所述的s-2-氨基丁酰胺可以以游离碱的形式使用或者以其盐的形式使用。s-2-氨基丁酰胺盐的形式可以为s-2-氨基丁酰胺盐酸盐。
[0025]
本发明中,所述的有机溶剂优选醚类溶剂。所述的醚类溶剂优选四氢呋喃。
[0026]
本发明中,所述的有机溶剂与所述的化合物iii的体积质量比值优选1ml/g~100ml/g,进一步优选5ml/g~20ml/g,例如13ml/g、15ml/g或17ml/g。
[0027]
本发明中,所述的s-2-氨基丁酰胺与所述的化合物iii的摩尔比值优选1.0~4.0,进一步优选2.0~3.0,例如2.5或2.2。
[0028]
本发明中,所述的缩合反应的温度优选-35℃~25℃,进一步优选-20℃~10℃,例如0℃~5℃或-10℃~-5℃。
[0029]
本发明中,所述的缩合反应的时间可以采用本领中的常规检测方法(例如tlc、hplc或nmr)进行监测,一般以所述的化合物iii消失时为反应的终点,所述的缩合反应的时间优选0.5小时~5小时,进一步优选0.5小时~2小时,例如1小时。
[0030]
本发明中,所述的缩合反应结束后,不进行化合物ii分离提纯直接进行亲核取代反应,制备得到布立西坦i。
[0031]
本发明中,所述的碱优选氢氧化钠、氢氧化钾、叔丁醇钠、叔丁醇钾、碳酸铯、氢化钠和氢氧化锂中的一种或多种。
[0032]
本发明中,所述的碱与所述的化合物iii的摩尔比值优选1.0~5.0,进一步优选2.0~3.5,例如3.0、2.5或2.2。
[0033]
本发明中,所述的亲核取代反应的温度优选-35℃~25℃,进一步优选-20℃~10℃,例如0℃~5℃、-15℃~-5℃或-10℃~-5℃。
[0034]
本发明中,所述的亲核取代反应的时间可以采用本领中的常规检测方法(例如
tlc、hplc或nmr)进行监测,一般以所述的中间体化合物ii消失时为反应的终点,所述的亲核取代反应的时间优选0.5小时~5小时,进一步优选0.5小时~2小时,例如1小时。
[0035]
本发明所述的布立西坦的制备方法优选包括以下步骤:将化合物iii与有机溶剂形成的溶液加入到s-2-氨基丁酰胺与有机溶剂形成的混合物中,进行缩合反应;然后,再加入碱与有机溶剂形成的混合物,进行亲核取代反应,得到所述的布立西坦i即可。
[0036]
所述的加入的方式优选滴加,所述的滴加的速度以体系温度不超过30℃为准,优选不超过25℃。
[0037]
本发明所述的布立西坦的制备方法优选包括以下后处理步骤:反应结束后,过滤、除去溶剂得到布立西坦i的粗品。所述的布立西坦i的粗品优选经过重结晶或者打浆得到布立西坦i。
[0038]
所述的重结晶优选包括以下步骤:将布立西坦i粗品与溶剂形成的溶液与活性炭混合搅拌,然后热滤,降温析晶,得到纯化后的布立西坦i即可。
[0039]
所述的混合搅拌的温度优选60℃~80℃,例如70℃。所述的混合搅拌的时间优选0.5小时~2小时,例如1小时。所述的降温析晶的温度优选0℃-30℃,例如15℃~25℃。所述的降温析晶的时间优选1小时~5小时,例如2小时或3小时。
[0040]
所述的重结晶采用的溶剂优选极性非质子有机溶剂,或,极性非质子有机溶剂与脂肪醇类溶剂的混合溶剂。
[0041]
所述的极性非质子有机溶剂优选酯类溶剂、醚类溶剂、酮类溶剂和腈类溶剂中的一种或多种。所述的酯类溶剂优选乙酸乙酯和/或乙酸异丙酯。所述的醚类溶剂优选四氢呋喃和/或异丙醚;所述的酮类溶剂优选丙酮和/或甲基异丁基酮。所述的腈类溶剂优选乙腈。所述的脂肪醇类溶剂优选异丙醇、甲醇和乙醇中的一种或多种。
[0042]
所述的打浆采用的溶剂优选醚类溶剂;所述的醚类溶剂优选异丙醚。
[0043]
所述的布立西坦i的制备方法优选进一步包括以下步骤:极性非质子有机溶剂中,将化合物iv与溴化试剂进行亲核取代反应,得到所述的化合物iii即可;
[0044][0045]
化合物iii的制备方法,可以在惰性气体保护下进行;所述的惰性气体优选氮气和/或氩气。
[0046]
化合物iii的制备方法可以在无溶剂或者极性非质子有机溶剂中进行,当化合物iii的制备在剂型非质子有机溶剂中进行时,所述的极性非质子有机溶剂优选醚类溶剂、烷烃类溶剂、芳烃类溶剂和腈类溶剂中的一种或多种。所述的醚类溶剂优选四氢呋喃和/或2-甲基呋喃。所述的烷烃类溶剂优选正庚烷和/或正己烷。所述的芳烃类溶剂优选正庚烷、甲苯、苯和溴苯中的一种或多种。所述的腈类溶剂优选乙腈。所述的极性非质子有机溶剂与所述的化合物iv的体积质量比值优选1ml/g~15ml/g,进一步优选2ml/g~5ml/g,例如3ml/g。
[0047]
化合物iii的制备方法中,所述的溴化试剂优选二溴亚砜、溴化氢乙酸溶液、乙酰溴和三溴化磷中的一种或多种。
[0048]
化合物iii的制备方法中,所述的溴化试剂与所述的化合物iv的摩尔比值优选1.0~5.0,进一步优选1.1~3.0,例如1.3或2.0。
[0049]
化合物iii的制备方法中,所述的亲核取代反应的温度优选40℃~100℃,进一步优选50℃~80℃,例如65℃~70℃。
[0050]
化合物iii的制备方法中,所述的亲核取代反应的时间可以采用本领域的常规检测方法(例如hplc、tlc或nmr)进行检测,一般以所述的化合物iv消失时为反应的终点,所述的亲核取代反应的时间优选3小时~30小时,进一步优选10小时~20小时,例如16小时。
[0051]
化合物iii的制备方法优选采用以下步骤:惰性气体保护下,将溴代剂加入到化合物iv与有机溶剂形成的混合物中,进行亲核取代反应得到所述的化合物iii即可。
[0052]
化合物iii的制备方法优选采用以下后处理步骤:反应结束后,除去溶剂,得到所述的化合物iii。所述的除去溶剂优选采用减压蒸馏的方式。所述的减压蒸馏的温度优选40℃~100℃,进一步优选60℃~80℃,例如60℃~70℃。所述的减压蒸馏的真空度小于0.7mpa,进一步优选小于0.9mpa。
[0053]
本发明制得化合物iii之后,无需进一步纯化,直接用于布立西坦的制备。
[0054]
所述的布立西坦i的制备方法优选采用以下合成路线:
[0055][0056]
本发明中所述原料或试剂除特别说明之外,均市售可得。
[0057]
本发明中,所述的室温是指环境温度,为10℃~35℃。
[0058]
本发明的积极进步效果在于:本发明的制备方法将两步反应一锅法制得布立西坦,无须分离提纯中间产物化合物ii(化合物ii稳定性差),合成路线短,总摩尔收率高达到62%;后处理步骤和纯化方法简单,通过重结晶或者打浆而不是手性高压液相制备柱进行进一步纯化,可以制得布立西坦i化学纯度99.80%以上,其他单杂小于0.1%,且手性纯度高ee值大于99.80%,达到原料药(api)级别,适合于工业化生产。
具体实施方式
[0059]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0060]
实施例1:
[0061][0062]
室温下,在5l反应釜中加入1.5l正庚烷、500g(3.90mol,1.0eq)化合物iv,加入
1373g三溴化磷(5.07mol),氮气保护下,升温65℃~70℃反应16h,反应确认完毕后,氮气氛围下降至20℃-30℃以内,静置5min,取出上清液,60℃减压浓缩至冷凝器无回流,得到约1216g,冷却至20℃-30℃以内,降温转移物料备用,收率98%以上,hplc纯度95%。
[0063]
实施例2
[0064][0065]
室温下(20℃~25℃),向30l反应釜中加入16l四氢呋喃和1347gs-2-氨基丁酰胺盐酸盐(9.75mol,2.2eq),加毕搅拌均匀,水分控制小于0.1%,氮气置换3次,搅拌降温至0℃~5℃,取化合物iii 1216g(4.47mol,1.0eq)和2l四氢呋喃混合1小时后滴加,加毕0℃~5℃保温反应1h,另取叔丁醇钾1094g(9.75mol,2.2eq)分少量多次加入,加毕0℃~5℃保温反应1h,取乙酸398g(6.63mol)在0℃~5℃下滴入反应液中,搅拌2h,过滤,滤饼弃去,滤液加入6l水,搅拌均匀,减压浓缩去四氢呋喃,浓缩物加入二氯甲烷4l搅拌萃取,静置分层,下层有机相转至浓缩釜,50-60℃减压浓缩滤液至基本无滴下,浓缩物加入4l异丙醚升温70℃溶清,缓慢降温至析出,冰水降温过滤,滤饼用250ml冰异丙醚淋洗,得到布立西坦i粗品530g,摩尔收率64%,hplc纯度99.80%。
[0066]
实施例3:
[0067]
将布立西坦i粗品530g加入1.75l异丙醚搅拌,升温,溶清加入活性炭5g,70℃搅拌1h,热过滤,滤液降温至50℃-56℃搅拌,大量固体析出后50℃搅拌1h,缓慢降温至15℃-20℃,约2h,过滤,滤饼用异丙醚淋洗,固体湿品504g放入40℃真空干燥16h得到495g布立西坦i,hplc纯度99.93%,总摩尔收率59.77%(以化合物iii为起始原料计两步反应的总收率),ee值99.80%,其他最大单杂0.08%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1