一种依帕司他晶型B的结晶制备方法与流程
一种依帕司他晶型b的结晶制备方法
技术领域
1.本发明属于晶型药物制备技术领域,具体涉及一种依帕司他晶型b的结晶制备方法。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.依帕司他(epalrestat,cas号:82159-09-9),分子式为c
15h13
no3s2,分子量为319.40,化学结构式如下表所示。依帕司他作为一类醛糖还原酶抑制剂,可用于治疗糖尿病引发的末梢神经障碍、心跳异常等症状,最早由日本小野公司开发并上市。
[0004][0005]
依帕司他晶型a水溶性较差,导致体内溶解、释放量低,进而影响药物的生物利用度。多晶型作为药物普遍存在的一种现象,对改善药物理化性质具有重要的作用。同种药物的不同晶型往往具有不同的溶解性、稳定性等指标,进而会影响制剂产品的临床疗效。相关研究表明依帕司他不仅具有多晶型现象,而且还可与其他组分形成共晶产品。研究人员通过大量研究,发现并制备了依帕司他其他晶型产品。其中,专利jp2005298424a公开了依帕司他晶型b的制备方法,但由于其制备过程中溶剂用量极大,晶型b的制备效率极低,难以满足工业化的实际需求。专利cn105272934a提供了一种依帕司他晶型c的制备方法,通过甲基丁酮、甲基异丁酮与甲酸、乙酸的两次精制过程可获得纯度在99.9%以上的晶型c产品。由于晶型c制备溶剂沸点较高容易导致产品中溶剂残留超标,且晶型c缺乏临床数据,其应用推广受到很大限制。专利cn113277962a通过采用丙酮水溶剂体系成功制备了依帕司他-二甲双胍盐水合物,大幅提升了依帕司他的溶出速率,同时降低了二甲双胍的引湿性,然而该新盐型化合物目前仍处于临床前研究阶段,最终能否成功上市仍存在诸多不确定性。
[0006]
依帕司他晶型b作为依帕司他已经商业化生产的重要晶型产品,其制备仍存在诸多缺陷,在一定程度上限制了其的规模化应用。专利cn107629021a通过采用甲醇与四氢呋喃混合溶剂体系,使得晶型b制备所需的溶剂用量降低至 14倍,但由于四氢呋喃具有一定的生殖毒性,在使用过程中会给操作人员带来一定的危害,限制了该方法的应用与推广。专利cn108191788a通过使用甲醇、乙醇等低碳醇进行高温打浆操作获得依帕司他晶型b产品。由于该法在高温区间停留时间较长,导致过程收率较低,不适于工业化放大。虽然通过加入酸或碱类试剂可提高其过程收率,但酸或碱类试剂的加入可能会导致局部ph变化剧烈引起杂质含量增加,对产品质量稳定性带来极大的不确定性。因此,寻找一种便于工业化应用、过程收率高、质量稳定的依帕司他晶型b的结晶制备方法显得尤为重要。
技术实现要素:
[0007]
基于上述技术背景,本发明目的在于提供一种稳定高效制备依帕司他晶型b 的方法,为了实现该技术目的,本发明提供以下技术方案:
[0008]
本发明第一方面,提供一种依帕司他晶型b的结晶制备方法,所述制备方法包括如下步骤:
[0009]
将依帕司他原料溶于醇类溶剂中,溶解后溶液中固液比为0.01g/g~0.05g/g;将所述溶液转移至结晶器中进行一段蒸发,待馏出液达到所述醇类溶剂质量的 20~30%时停止蒸发,在此时温度终点恒温养晶30~60min;
[0010]
进行二段蒸发,待两次蒸发的馏出液总重量达到所述醇类溶剂质量的45~ 65%时,停止蒸发;
[0011]
降温至0~5℃,降温速率为1~2℃/min,并养晶60~90min;
[0012]
过滤结晶器中的反应体系,得到固体部分即为所述依帕司他晶型b。
[0013]
上述制备方法中,作为原料的依帕司他包括晶体形式或主要为晶体形式的依帕司他原料;优选的实施方式中,所述制备方法采用依帕司他晶型a和/或无定型依帕司他作为制备原料;
[0014]
一种的实施方式中,所述依帕司他原料为依帕司他晶型a,或晶型a含量为60%及以上;
[0015]
又一种的实施方式中,所述依帕司他原料为无定型依帕司他,或无定型含量为60%及以上;
[0016]
又一种的实施方式中,所述依帕司他原料中,晶型a及无定型态依帕司他含量为60%及以上。
[0017]
优选的,所述醇类溶剂为包括但不限于甲醇、乙醇、正丙醇、异丙醇中的一种或几种的混合溶剂。
[0018]
优选的,上述依帕司他原料加入醇类溶剂后,在60~70℃下连续搅拌溶解 30~60min得到所述溶液。
[0019]
优选的,所述一段蒸发采用真空蒸发结晶的方式,真空度为0.06~0.08mpa。
[0020]
优选的,所述二段蒸发同样采用真空蒸发结晶,真空度为0.04~0.06mpa。
[0021]
优选的,上述制备方法中,对结晶器内反应体系进行过滤后,还包括对所述固体部分进行洗涤及干燥的步骤。
[0022]
进一步的,所述洗涤采用的试剂选择易挥发且成本较低的有机试剂即可,干燥过程中即可完全去除,本发明验证可行的洗涤试剂为包括但不限于甲醇、乙醇、正丙醇、异丙醇中的一种。
[0023]
进一步的,所述干燥方式优选采用低温干燥,如真空干燥的方式,干燥温度为40~50℃,真空度为0.08~0.1mpa,干燥时间7~12小时。
[0024]
以上一个或多个技术方案的有益效果是:
[0025]
1、为了克服现有技术的不足,本发明针对依帕司他的结晶工艺进行了系统性研究,采用蒸发-冷却耦合结晶技术将其他晶型产品稳定转化为晶型b,产品的收率在90%以上,并且纯度能够达到99.9%以上。同时本发明得到的依帕司他晶体产品不聚集,粒度分布均匀,批次间质量重现性好。
[0026]
2、本发明提供的制备工艺过程简便,仅需要结晶器及低碳醇类试剂即可完成,适合工业放大生产。
附图说明
[0027]
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
[0028]
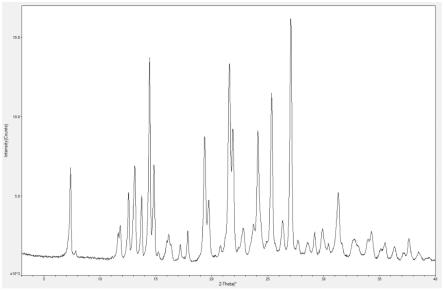
图1为实施例1中产品的xrd衍射图谱。
具体实施方式
[0029]
应该指出,以下详细说明都是例示性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
[0030]
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
[0031]
为了使得本领域技术人员能够更加清楚地了解本发明的技术方案,以下将结合具体的实施例详细说明本发明的技术方案。
[0032]
实施例1
[0033]
将10g依帕司他晶型a加入盛有500g甲醇溶剂的三口瓶中,在60℃下搅拌溶解,连续搅拌60分钟后,过滤,脱色;将滤液移入结晶器中,保持溶液温度为30℃,开启真空泵进行蒸发结晶操作,控制体系真空度为0.06mpa,当馏出液达到150g时,停止蒸发溶剂,恒温养晶搅拌30分钟;接着控制体系真空度为 0.04mpa,继续蒸发溶剂,当馏出液总量为325g时,停止蒸发操作;接着以2℃ /min的速率将溶液体系温度降至5℃,之后养晶60min。抽滤,并用甲醇洗涤滤饼,在40℃真空条件下进行干燥7小时,真空度为0.08mpa。最终产品收率为 92.3%,hplc纯度为99.92%,产品粒度d
50
为8.76微米。
[0034]
本实施例中产品的xrd衍射结果如附图1所示,与b晶型对照品衍射图进行比对,两者具有基本相同的特征衍射峰,可以确定上述产品中为b晶型产品。下述实施例中所述制备的产品采用同样的方式进行比对,可确认为b晶型产品。
[0035]
实施例2
[0036]
将10g无定型依帕司他加入盛有1000g乙醇溶剂的三口瓶中,在70℃下搅拌溶解,连续搅拌30分钟后,过滤,脱色;将滤液移入结晶器中,保持溶液温度为40℃,开启真空泵进行蒸发结晶操作,控制体系真空度为0.08mpa,当馏出液达到300g时,停止蒸发溶剂,恒温养晶搅拌60分钟;接着控制体系真空度为0.06mpa,继续蒸发溶剂,当馏出液总量为500g时,停止蒸发操作;接着以 1℃/min的速率将溶液体系温度降至0℃,之后养晶90min。抽滤,并用乙醇洗涤滤饼,在50℃真空条件下进行干燥12小时,真空度为0.1mpa。最终产品收率为91.8%,hplc纯度为99.94%,产品粒度d
50
为7.85微米。
[0037]
实施例3
[0038]
将20g依帕司他晶型a加入盛有1000g异丙醇溶剂的三口瓶中,在65℃下搅拌溶解,
连续搅拌50分钟后,过滤,脱色;将滤液移入结晶器中,保持溶液温度为40℃,开启真空泵进行蒸发结晶操作,控制体系真空度为0.07mpa,当馏出液达到200g时,停止蒸发溶剂,恒温养晶搅拌45分钟;接着控制体系真空度为0.05mpa,继续蒸发溶剂,当馏出液总量为650g时,停止蒸发操作;接着以1℃/min的速率将溶液体系温度降至2℃,之后养晶70min。抽滤,并用异丙醇洗涤滤饼,在55℃真空条件下进行干燥10小时,真空度为0.09mpa。最终产品收率为90.3%,hplc纯度为99.93%,产品粒度d
50
为6.56微米。
[0039]
实施例4
[0040]
将10g依帕司他晶型a加入盛有250g正丙醇溶剂的三口瓶中,在70℃下搅拌溶解,连续搅拌45分钟后,过滤,脱色;将滤液移入结晶器中,保持溶液温度为35℃,开启真空泵进行蒸发结晶操作,控制体系真空度为0.08mpa,当馏出液达到50g时,停止蒸发溶剂,恒温养晶搅拌30分钟;接着控制体系真空度为0.06mpa,继续蒸发溶剂,当馏出液总量为125g时,停止蒸发操作;接着以 1℃/min的速率将溶液体系温度降至1℃,之后养晶80min。抽滤,并用正丙醇洗涤滤饼,在50℃真空条件下进行干燥9小时,真空度为0.1mpa。最终产品收率为91.7%,hplc纯度为99.94%,产品粒度d
50
为9.21微米。
[0041]
实施例5
[0042]
将10g无定型依帕司他加入盛有400g甲醇溶剂的三口瓶中,在65℃下搅拌溶解,连续搅拌60分钟后,过滤,脱色;将滤液移入结晶器中,保持溶液温度为30℃,开启真空泵进行蒸发结晶操作,控制体系真空度为0.06mpa,当馏出液达到40g时,停止蒸发溶剂,恒温养晶搅拌50分钟;接着控制体系真空度为 0.05mpa,继续蒸发溶剂,当馏出液总量为180g时,停止蒸发操作;接着以2℃ /min的速率将溶液体系温度降至0℃,之后养晶90min。抽滤,并用甲醇洗涤滤饼,在40℃真空条件下进行干燥8小时,真空度为0.09mpa。最终产品收率为 90.6%,hplc纯度为99.94%,产品粒度d
50
为9.64微米。
[0043]
实施例6
[0044]
将20g无定型依帕司他加入盛有500g乙醇溶剂的三口瓶中,在70℃下搅拌溶解,连续搅拌45分钟后,过滤,脱色;将滤液移入结晶器中,保持溶液温度为30℃,开启真空泵进行蒸发结晶操作,控制体系真空度为0.07mpa,当馏出液达到125g时,停止蒸发溶剂,恒温养晶搅拌40分钟;接着控制体系真空度为 0.05mpa,继续蒸发溶剂,当馏出液总量为250g时,停止蒸发操作;接着以1.5℃ /min的速率将溶液体系温度降至5℃,之后养晶60min。抽滤,并用乙醇洗涤滤饼,在45℃真空条件下进行干燥12小时,真空度为0.09mpa。最终产品收率为 90.2%,hplc纯度为99.93%,产品粒度d
50
为8.63微米。
[0045]
实施例7
[0046]
将20g无定型依帕司他加入盛有500g异丙醇溶剂的三口瓶中,在60℃下搅拌溶解,连续搅拌40分钟后,过滤,脱色;将滤液移入结晶器中,保持溶液温度为35℃,开启真空泵进行蒸发结晶操作,控制体系真空度为0.08mpa,当馏出液达到150g时,停止蒸发溶剂,恒温养晶搅拌30分钟;接着控制体系真空度为0.04mpa,继续蒸发溶剂,当馏出液总量为300g时,停止蒸发操作;接着以2℃/min的速率将溶液体系温度降至0℃,之后养晶60min。抽滤,并用异丙醇洗涤滤饼,在50℃真空条件下进行干燥12小时,真空度为0.08mpa。最终产品收率为92.6%,hplc纯度为99.92%,产品粒度d
50
为7.53微米。
[0047]
实施例8
[0048]
将10g依帕司他晶型a加入盛有200g正丙醇溶剂的三口瓶中,在70℃下搅拌溶解,连续搅拌60分钟后,过滤,脱色;将滤液移入结晶器中,保持溶液温度为40℃,开启真空泵进行蒸发结晶操作,控制体系真空度为0.07mpa,当馏出液达到60g时,停止蒸发溶剂,恒温养晶搅拌40分钟;接着控制体系真空度为0.05mpa,继续蒸发溶剂,当馏出液总量为110g时,停止蒸发操作;接着以2℃ /min的速率将溶液体系温度降至5℃,之后养晶70min。抽滤,并用正丙醇洗涤滤饼,在55℃真空条件下进行干燥10小时,真空度为0.09mpa。最终产品收率为92.1%,hplc纯度为99.91%,产品粒度d
50
为8.46微米。
[0049]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1